METHOD DEVELOPMENT
COLUMN SCREENING
Before a target compound can be isolated chromatographically, method development is required. This is typically done on the analytical scale in order to save solvent, time and sample. In SFC, the retention of single components of a mixture on a defined stationary phase cannot be predicted. Unlike RPLC, the identification of the right column chemistry is critical for SFC separations and is the most important factor in SFC method development. It is, therefore, important to rely on an efficient screening strategy to maximize the quality of the separation with respect to the target(s) of the purification.7 Stationary phase development is currently very dynamic, and a small number of chemistries could emerge as the preferred columns in the future. Today, most users adapt an automated screening process using the “best” columns for their application.7
Several factors need to be considered to determine an appropriate set of columns for screening. L/dp (length/particle size) needs to be maintained between the analytical column and the eventual preparative column, so only appropriately-sized analytical columns should be considered. This ensures continuity of the chromatography between systems on scale-up. The columns should have a wide range of selectivity, and also be appropriate for the nature of the sample. For example, highly polar compounds (like carbohydrates) are not likely to retain on a C18 column, while a silica column is probably not optimal for extremely hydrophobic (non-polar) compounds (like most carotenoids). Columns can also be classified according to their suitability for basic or acidic compounds, or both.
For chiral purification, chiral columns are required. Due to the more complex stationary phase interaction with target compounds, more trial and error is required to determine the best column for chiral separations. However, some columns have a higher hit rate than others. For example, cellulose and amylose derived chiral phases are popular
for their wide range of applicability,7 while other chiral phases, such as the pirkle type phases are popular for their high chemical stability. Chiral phases can also be used successfully for achiral applications, especially with positional isomers or closely related compounds.3
Column screening is typically done using gradient solvent conditions, usually between 2% and 50% co-solvent. These conditions give the user a good idea of which column will be optimal for isolating the target compound, while also providing a good indication of what co-solvent percentages are required for compound elution. Figure 17 is an example chiral column screen for the separation of (R) and (S)-goitrin (an active natural product found in Isatis Indigotica Fort root), using five cellulose and amylose based stationary phases (IC, OJ-H, AS-H, OD-H, and AD-H), and one pirkle type stationary phase (S,S)-Whelk-O1.16 An example achiral column screening can be seen in Figure 18, where a mixture of three carotenoids is screened on four achiral stationary phases.17 Notice the carotenoids are best retained on the C18, and not on the other more polar phases.
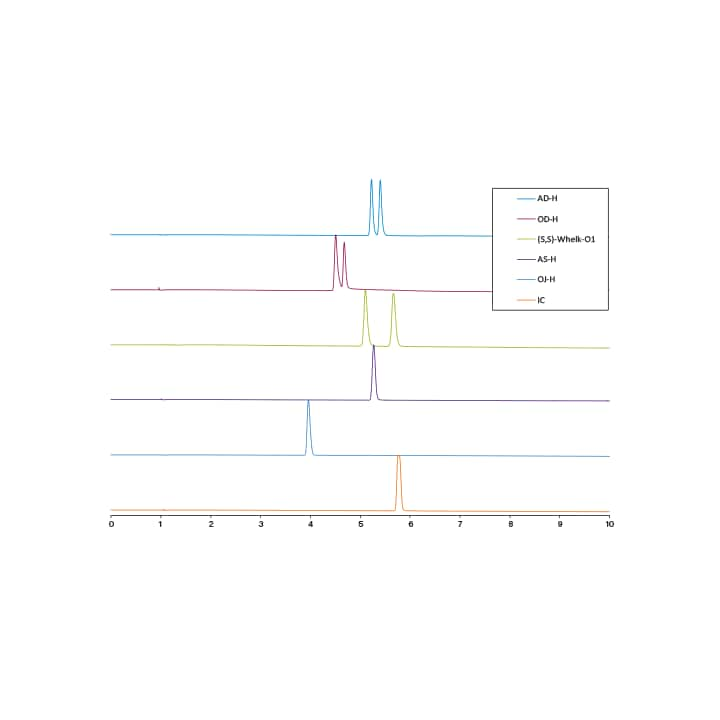
Figure 17. Example of a chiral column screen. SFC Chromatograms of the (R,S)-goitrin standard run on six different stationary phases. Gradient was as follows: 5–40% 10 min, hold at 40% 2 min, 40–5% 1 min, hold at 5% 2 min.16
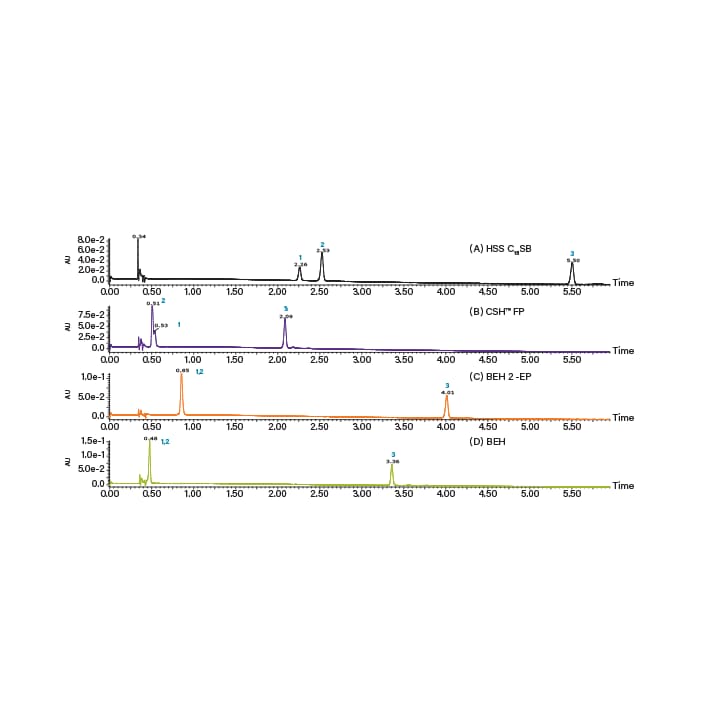
Figure 18. Example of an achiral column screen. UPC2 UV chromatograms of lycopene, ß-carotene and lutein mixture obtained using different columns: (A) HSS C18 SB; (B) CSH Fluoro-Phenyl; (C) BEH 2-EP; and (D) BEH. The identities of the peaks are: 1. Lycopene; 2. ß-carotene; and 3. Lutein. Gradient was as follows: 5–20% in 5 min, hold at 20% for 2 min, 20–5% in 1 min.17
SOLVENT SCREENING
CO2 alone is generally insufficient for elution of compounds from a chromatographic column; as such, a polar co-solvent is usually added. Co-solvent selection is a key parameter in SFC chromatographic method development and optimization. For most applications, a wide range of co-solvents and mixtures can be applied to optimize the separation. The range of compatible solvents in SFC can seem overwhelming, however it can be simplified by pairing both ends of the polarity spectrum; non-polar CO2 with highly polar organic solvents, resulting in a mobile phase that covers a large range of solvent strength.
The four most typically used co-solvents are methanol, ethanol, isopropanol and acetonitrile; methanol being the strongest and acetonitrile being the weakest in polarity. Usually, methanol or ethanol is recommended for the column screen and initial solvent screen. Isopropanol and acetonitrile are good for optimizing the method or in cases where ethanol or methanol do not result in acceptable resolution. Many times, combinations of these solvents can fine tune the polarity of the mobile phase. For example, adding acetonitrile to ethanol weakens the co-solvent, increasing retention and providing a different selectivity. Usually as the co-solvent strength decreases, retention increases (Figure 19). Also, just like in LC, as the proportion of strong solvent increases retention decreases (Figure 20). Sometimes however, due to the low viscosity of the CO2, the total flow rate can be increased (at the same co-solvent percentage) resulting in a faster run time while still maintaining the separation (Figure 21).
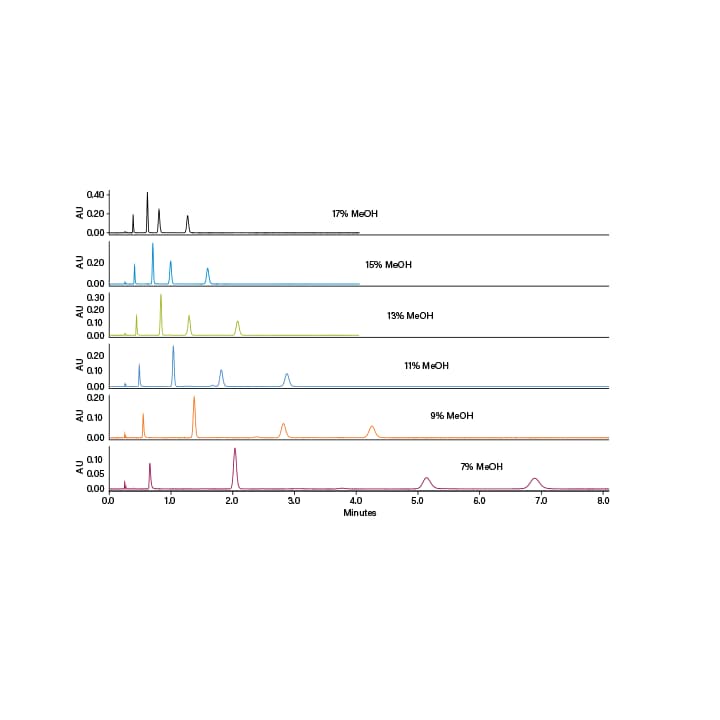
Figure 20. Effect of co-solvent percentage on retention.
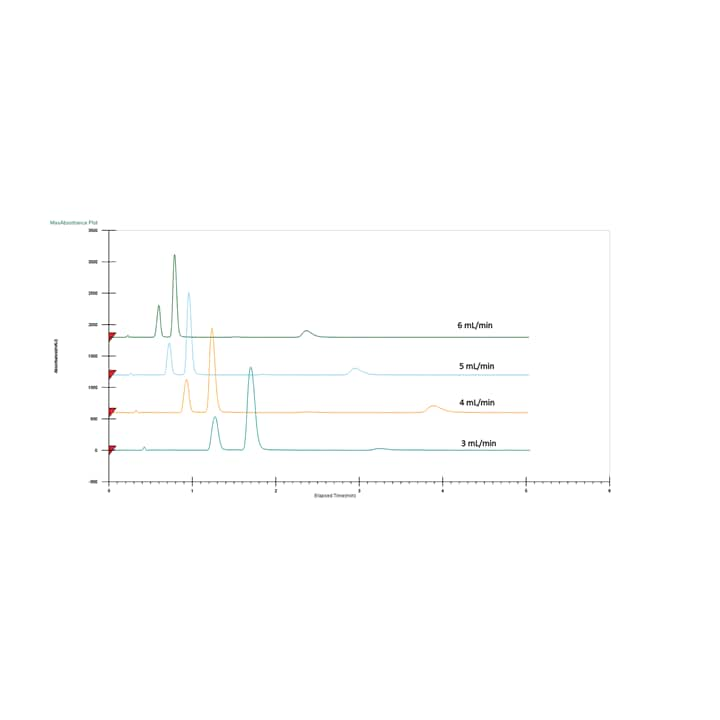
Figure 21. Effect of total flow rate on chromatography.
ADDITIVES IN THE CO-SOLVENT
Sharper peaks and improved resolution are important in prep chromatography, where broad peaks and poor solubility can have disastrous results for productivity. Peak tailing in SFC can be suppressed with the use of additives in the co-solvent portion of the mobile-phase, which extends the range of solutes amenable to a CO2 based mobile phase.10
Utilizing CO2 as a chromatographic solvent, results in a slightly acidic mobile phase when paired with other polar organic solvents. Because a CO2 based mobile phase is slightly acidic, many acidic and neutral compounds exhibit acceptable peak shapes without the need for a mobile phase additive. However, acidic additives can improve peak shapes for some acidic compounds (Figure 22). Common acidic additives include trifluoroacetic acid, formic acid, and acetic acid. Applications containing basic compounds typically require a basic additive (usually between 0.1 and 1%) to improve the chromatography (Figure 23). Common basic additives are secondary or tertiary amines such as isopropylamine (IPA), diethylamine (DEA) or triethylamine (TEA).These additives are extremely effective even in very low concentrations (as low as 0.01% in the co-solvent), however they can interfere with MS detection and may present memory effects on some column chemistries (specifically bare silica). As a result, ammonium acetate and ammonium hydroxide are used as well, since they are more column and MS friendly, and can even enhance MS signal.
In addition to the classical co-solvents, many new columns are compatible with non-classical co-solvents or additives, including ethyl acetate, tetrahydrofuran, dichloromethane, chloroform or dimethoxymethane. These combinations permit
retention times and selectivity to be modulated even further, while improving sample solubility.⁷ One non-classical additive that is gaining popularity is water. Water is immiscible with CO2; however, when used as an additive in a strongly polar co- solvent (like methanol) water becomes an excellent choice for increasing the solubility and peak shape of hydrophilic compounds in SFC (Figure 24). The amount of water that can be tolerated is dependent on the proportion of the co-solvent in the total mobile phase. Eventually, baseline noise will be observed due to phase separation (immiscibility) in the mobile phase.10 Usually, 1% to 5% water can be used effectively in the polar co-solvent. With water as an additive, more strongly polar compounds can be purified by Prep SFC.³
For purification purposes, it is best to avoid the use of additives that will need to be removed downstream. If additives are necessary for the separation, volatile additives that can be easily removed should be used in the lowest effective concentration. Alternatively, there are now new stationary phases that reduce the need for additives all together, which provides a significant benefit to Prep SFC.
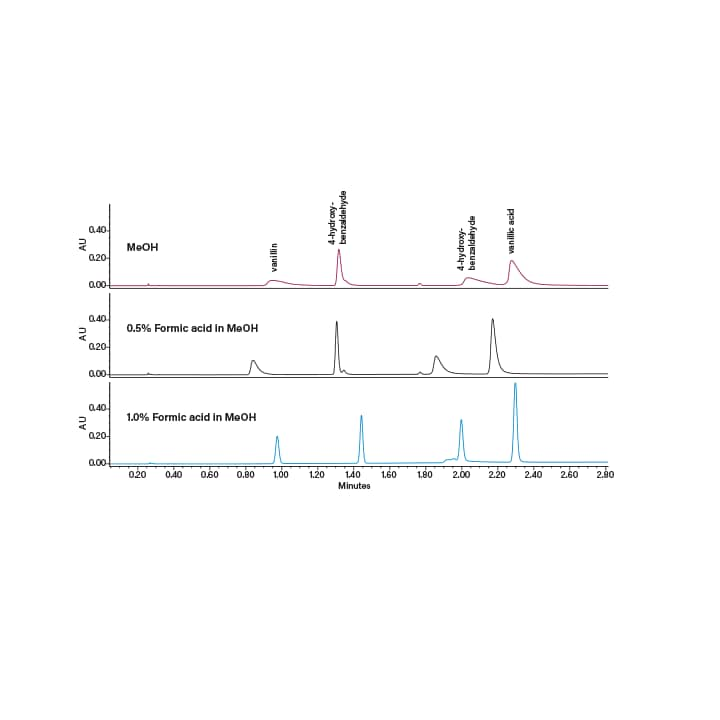
Figure 22. Effect of acidic additives on peak shape and resolution of acidic compounds.
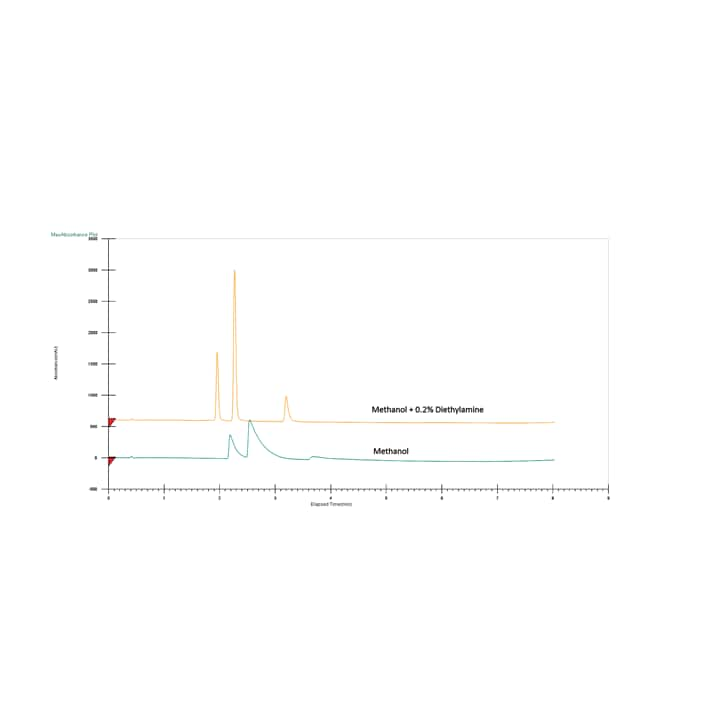
Figure 23. Effect of basic additive on peak shape and resolution of basic compounds.
DENSITY: TEMPERATURE AND PRESSURE
Solubility and retention factor of all compounds are closely related to the fluid density.
Mobile phase density along the column is of great importance because it controls the physical and chemical properties of the mobile phase.⁵ An interesting feature of SFC is the ability to control the density of the mobile phase using temperature and pressure. While the choice of column and mobile phase has the greatest impact on the separation in SFC, temperature and pressure are used to fine tune or optimize a separation. Between the two parameters, pressure has the greatest influence on the chromatography; as the pressure increases the density increases, typically resulting in a reduced retention time and resolution (Figure 25). Higher temperatures on the other hand result in lower density and longer retention times. With temperature however, the effect is compound dependent and can result in a change in resolution, or even elution order for closely eluting compounds; this has been observed more frequently for chiral separations (Figure 26).
The pressure is controlled using a back pressure regulator (BPR) that sets the system “back pressure” after the column. Although the systems are designed for a wide range of pressures (up to 400 bar set points), in Prep SFC, the back pressure regulator is typically set between 100 and 200 bar (1450–2901 psi). Temperatures are
controlled by set points either using an oven (heating the column) or heat exchangers (heating the mobile phase). Typical temperature set points are between 40 and 60 °C. Some applications are run at subcritical (liquid CO2) conditions, where the temperatures are set lower between 25 and 35°C, while the pressure is kept relatively high. It is not recommended to run at pressures and temperatures close to the critical point because at those conditions, small changes in temperature or pressure result in large changes in density (and chromatography).⁵ Both temperature and pressure limitations are typically related to the robustness of the column chemistry or packing.
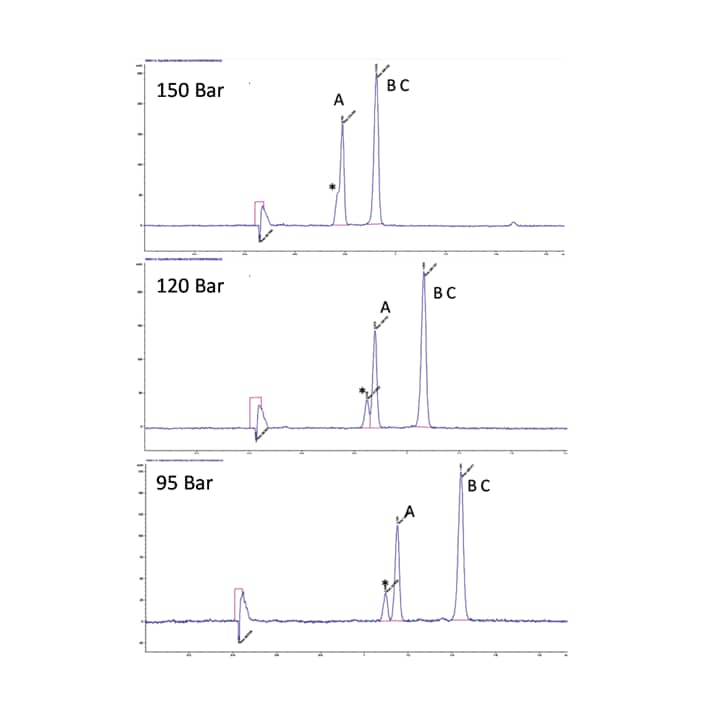
Figure 25. Effect of pressure on retention and resolution.
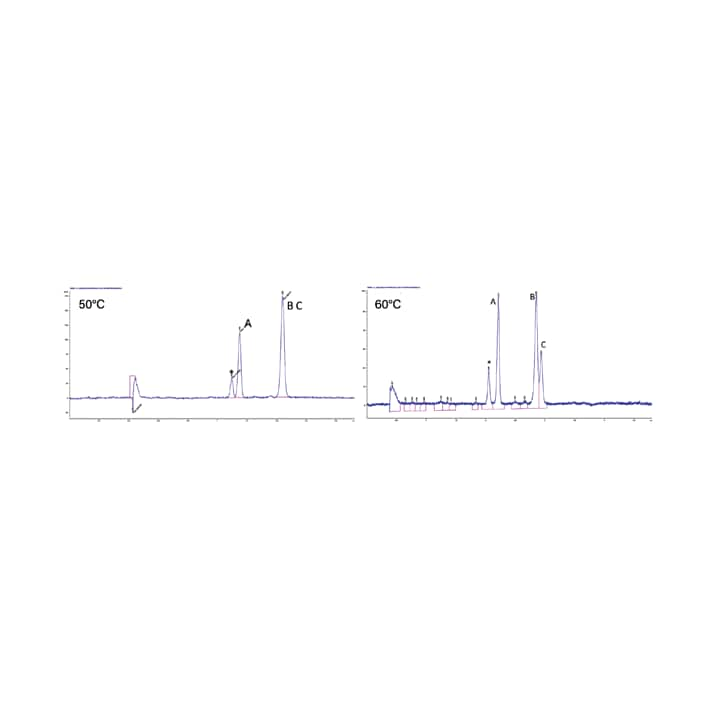
Figure 26. Effect of temperature on retention and resolution.
OPTIMIZING MOBILE PHASE CONDITIONS
Choosing the optimal combination of solvent and column is usually application or goal specific. In a perfect scenario, high loading, good resolution, and fast separation combine to provide complete recovery of the target at a high purity. In reality, however, compromise is usually necessary to determine the most productive solution. For example, the peaks can be well separated (allowing for high loading) but require a longer run time. On the other hand, a good separation with a significantly shorter run time would allow for a faster cycle time, but possibly with lower loading. Another consideration is down-stream processing or fraction recovery, where the amount or type of solvent can be an important factor. Finally, if the separation can be accomplished under isocratic conditions, stacked injections can be utilized, greatly improving productivity. Once a column and solvent are selected, other parameters can be manipulated to better optimize the separation before scale-up and purification.
Optimizing gradient conditions: In SFC, focused gradients do not work the same as in RPLC. Because of the many competing retention mechanisms in normal-phase chromatography, changes in the gradient can have disparate effects on the retention of different analytes. In SFC, specifically, there is also a change in density and pressure drop that accompanies the increase in co-solvent across the gradient. As the amount of co-solvent increases, the effect on retention time is not linear, making it difficult to predict the selectivity of the compounds with the changing conditions. For structurally similar compounds this is less of a problem, as they follow similar retention mechanisms. Samples that contain mixtures of compounds that are structurally different (in a matrix, for example) present a bigger challenge. In general, however, shallower gradients result in longer retention times and better resolution. Figure 27 shows an example of a focused gradient in SFC for an indicated target compound.
Determining isocratic conditions: Isocratic methods are ideal because they are easy to develop based on the screening results, and stacked injections can be used, improving productivity. Using retention time, slope of the screening gradient and compensating for system and column volume delay, the co-solvent percentages at elution can be determined. In SFC, usually the best starting point for optimization is 5% below the calculated percentage.
Using the separation in Figure 28 as an example, the screening gradient was 2 to 20% over 5 minutes and the gradient delay was 0.46 min (previously determined). So with a calculated slope of 3.6%/min and 2% starting percentage, the co-solvent percentage at elution of the first peak at 4.12 minutes was calculated using the following equation:
■ %Co-solvent at Elution = (retention time – gradient delay) x gradient slope + starting %
■ %Co-solvent at Elution = (4.12 min – 0.46 min) x 3.6%/min + 2%
■ %Co-solvent at Elution = 15%
Therefore, after subtracting 5%, 10% isocratic co-solvent conditions were used as a starting point for optimization. The resulting chromatography showed good separation; but by increasing the co-solvent fraction of the mobile phase back to 15%, the peaks were still well resolved with a shorter run time. In this case, the 10% method would allow for higher loading, while the 15% method would allow for faster cycle times.
CHOOSING A PREP SYSTEM
SYSTEM SCALE
For Prep SFC, instruments with the capacity to operate at flow rates of 2 mL/min to several hundreds of mL/min are available to meet the requirements needed to work at various scales.⁷ In a proper workflow, the scale of the purification needs to match the goal of the application. This is particularly important when the sample is limited, or has low solubility. In that situation, a smaller scale (semi-prep) purification system is more practical. In SFC, the small scale purifications are usually performed on columns with 4.6mm to 10mm ID, at flow rates from 3 to 20mL/min. A table matching column size with flow rates and approximate loading capacity in Prep SFC is shown below (Table 7). It is important to note that loading capacity for a particular sample is dependent on many factors including solubility, resolution of the target, and the relative quantity of target compound to the matrix.
|
Column ID
5 µm particles
|
Flow rate range
|
Loading (per injection)
|
|
4.6 mm
|
3–6 mL/min
|
Up to 1 mg
|
|
10 mm
|
10–20 mL/min
|
Up to 5 mg
|
|
19 mm
|
50–100 mL/min
|
Up to 100 mg
|
|
30 mm
|
100–200 mL/min
|
Up to 300 mg
|
|
50 mm
|
250–350 mL/min
|
Up to 800 mg
|
Table 7. Table of column IDs, flow rates, and estimated loading capacity per injection.
Larger scale Prep SFC systems utilize 18-50mm ID columns at flow rates from 50 to 350 mL/min. The larger column capacity allows for more loading per injection and higher flow rates, which optimizes throughput for those applications. In the all-encompassing world of purification, these systems are considered mid-scale. Large pilot-scale Prep SFC systems (not addressed here) are used in many established industrial processes. These use very large (30 cm ID or larger) high pressure dynamic axial compression (DAC) chromatography columns, and are typically operated as plant facilities.
WORKFLOWS: “BULK” OR “BATCH”
Depending on the application, there are two basic workflows utilized in Prep SFC. The first is referred to as “bulk” purification. In this workflow, a single large quantity sample is injected multiple times until the required quantity of the target(s) is recovered, or the sample is exhausted. All matching collected fractions are pooled into one of a limited number of collection bottles. “Bulk” amounts of material are processed and recovered over the course of several hours (or sometimes even days). Typically, this is UV directed purification, although other methods of detection can be utilized. In this case, usually the samples are well characterized and understood and high productivity is the goal.
For many “bulk” applications, stacked injections are used. Stacked injections significantly improve throughput by utilizing all of the available chromatographic space for continuous separation and purification. Essentially, stacked injections reduce the time between injection cycles and minimize solvent usage even further. An example of stacked injections can be seen in Figure 29. In some applications, several injections can be made before the first set of peaks has eluted. For stacked injections to be utilized, the system must be able to inject while collections are occurring. Therefore, a separate injection module or a system capable of autonomous movement of the collection and injection assemblies (needles and probes) is used.
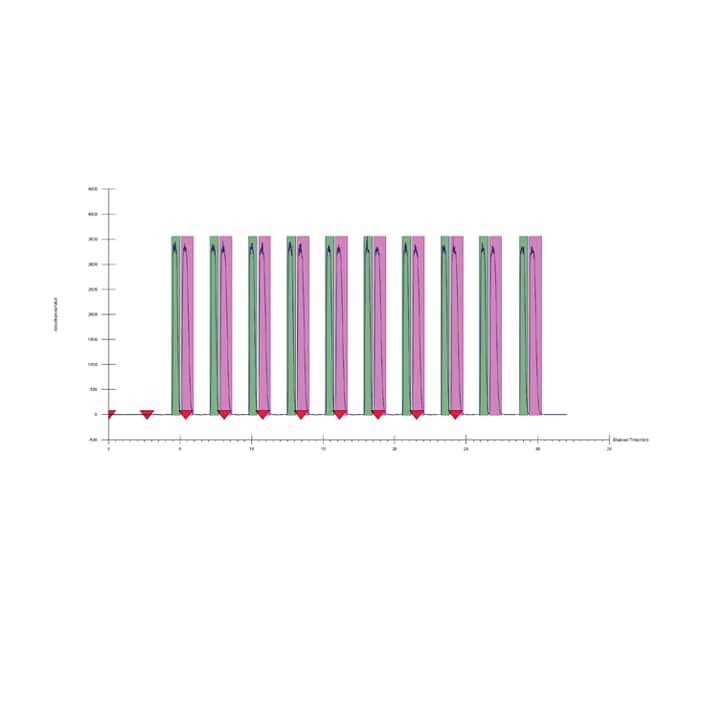
Figure 29: Stacked injections and collections of the enantiomers of flurbiprofen.
“Batch” purification refers to a workflow that is most practical for library purification, or when there are multiple samples with multiple targets that need to be purified. Usually, mass-directed purification is used because it is more selective when collecting fractions. With optical (or UV) directed purification, the peaks cannot be distinguished from one another by the detector. Many compounds absorb at the same wavelength. Mass-directed purification collects fractions based on mass, which is a much more specific parameter because it distinguishes between the target(s) and any impurities. This is especially useful when the samples have complex matrices, are not well characterized, or cannot be detected by UV because they do not have chromophores. Typically, gradient methods are used to improve peak shape and better separate compounds from complex interferences. This workflow utilizes open bed collection into multiple tubes; typically each tube constitutes a single fraction. The samples are run as a “batch”, and target compounds are collected based on their masses. Batch purification can also be done using UV-only or UV and MS triggers. Example MS based purification of target compounds from natural products can be seen in Figure 30.
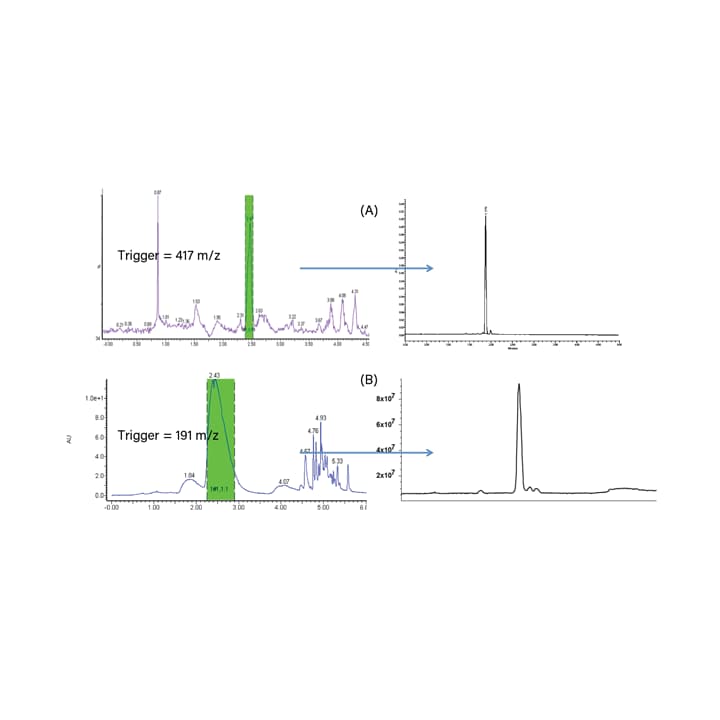
Figure 30. MS based purification of target compounds from natural products, including: (A) Schisandra berry extract and (B) Dang Gui root extract.18,19
