In general, three primary characteristics of chemical compounds can be used to create HPLC separations. They are:
• Polarity
• Electrical Charge
• Molecular Size
First, let’s consider polarity and the two primary separation modes that exploit this characteristic: normal phase and reversed-phase chromatography.
Separations Based on Polarity
A molecule’s structure, activity, and physicochemical characteristics are determined by the arrangement of its constituent atoms and the bonds between them. Within a molecule, a specific arrangement of certain atoms that is responsible for special properties and predictable chemical reactions is called a functional group. This structure often determines whether the molecule is polar or non-polar. Organic molecules are sorted into classes according to the principal functional group(s) each contains. Using a separation mode based on polarity, the relative chromatographic retention of different kinds of molecules is largely determined by the nature and location of these functional groups. As shown in Figure P, classes of molecules can be ordered by their relative retention into a range or spectrum of chromatographic polarity from highly polar to highly non-polar.
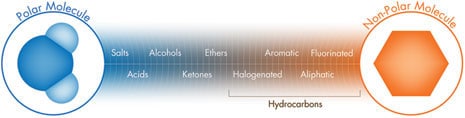
Figure P: Chromatographic Polarity Spectrum by Analyte Functional Group
Water [a small molecule with a high dipole moment] is a polar compound. Benzene [an aromatic hydrocarbon] is a non-polar compound. Molecules with similar chromatographic polarity tend to be attracted to each other; those with dissimilar polarity exhibit much weaker attraction, if any, and may even repel one another. This becomes the basis for chromatographic separation modes based on polarity.
Another way to think of this is by the familiar analogy: oil [non-polar] and water [polar] don’t mix. Unlike in magnetism where opposite poles attract each other, chromatographic separations based on polarity depend upon the stronger attraction between likes and the weaker attraction between opposites. Remember, “Like attracts like” in polarity-based chromatography.

Figure Q: Proper Combination of Mobile and Stationary Phases Effects Separation Based on Polarity
To design a chromatographic separation system [see Figure Q], we create competition for the various compounds contained in the sample by choosing a mobile phase and a stationary phase with different polarities. Then, compounds in the sample that are similar in polarity to the stationary phase [column packing material] will be delayed because they are more strongly attracted to the particles. Compounds whose polarity is similar to that of the mobile phase will be preferentially attracted to it and move faster.
In this way, based upon differences in the relative attraction of each compound for each phase, a separation is created by changing the speeds of the analytes.
Figures R-1, R-2, and R-3 display typical chromatographic polarity ranges for mobile phases, stationary phases, and sample analytes, respectively. Let’s consider each in turn to see how a chromatographer chooses the appropriate phases to develop the attraction competition needed to achieve a polarity-based HPLC separation.
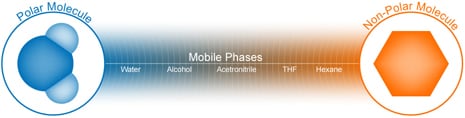
Figure R-1: Mobile Phase Chromatographic Polarity Spectrum
A scale, such as that shown in Figure R-1, upon which some common solvents are placed in order of relative chromatographic polarity is called an eluotropic series. Mobile phase molecules that compete effectively with analyte molecules for the attractive stationary phase sites displace these analytes, causing them to move faster through the column [weakly retained]. Water is at the polar end of mobile-phase-solvent scale, while hexane, an aliphatic hydrocarbon, is at the non-polar end. In between, single solvents, as well as miscible-solvent mixtures [blended in proportions appropriate to meet specific separation requirements], can be placed in order of elution strength. Which end of the scale represents the ‘strongest’ mobile phase depends upon the nature of the stationary phase surface where the competition for the analyte molecules occurs.

Figure R-2: Stationary Phase Particle Chromatographic Polarity Spectrum
Silica has an active, hydrophilic [water-loving] surface containing acidic silanol [silicon-containing analog of alcohol] functional groups. Consequently, it falls at the polar end of the stationary-phase scale shown in Figure R-2. The activity or polarity of the silica surface may be modified selectively by chemically bonding to it less polar functional groups [bonded phase]. Examples shown here include, in order of decreasing polarity, cyanopropylsilyl- [CN], n-octylsilyl- [C8], and n-octadecylsilyl- [C18, ODS] moieties on silica. The latter is a hydrophobic [water-hating], very non-polar packing.
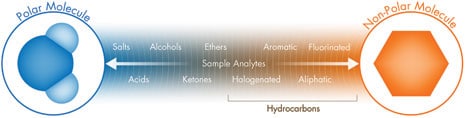
Figure R-3: Compound/Analyte Chromatographic Polarity Spectrum
Figure R-3 repeats the chromatographic polarity spectrum of our sample [shown in Figure P]. After considering the polarity of both phases, then, for a given stationary phase, a chromatographer must choose a mobile phase in which the analytes of interest are retained, but not so strongly that they cannot be eluted. Among solvents of similar strength, the chromatographer considers which phase combination may best exploit the more subtle differences in analyte polarity and solubility to maximize the selectivity of the chromatographic system. Like attracts like, but, as you probably can imagine from the discussion so far, creating a separation based upon polarity involves knowledge of the sample and experience with various kinds of analytes and retention modes. To summarize, the chromatographer will choose the best combination of a mobile phase and particle stationary phase with appropriately opposite polarities. Then, as the sample analytes move through the column, the rule like attracts like will determine which analytes slow down and which proceed at a faster speed.
Normal-Phase HPLC
In his separations of plant extracts, Tswett was successful using a polar stationary phase [chalk in a glass column; see Figure A] with a much less polar [non-polar] mobile phase. This classical mode of chromatography became known as normal phase.

Figure S-1: Normal-Phase Chromatography
Figure S-1 represents a normal-phase chromatographic separation of our three-dye test mixture. The stationary phase is polar and retains the polar yellow dye most strongly. The relatively non-polar blue dye is won in the retention competition by the mobile phase, a non-polar solvent, and elutes quickly. Since the blue dye is most like the mobile phase [both are non-polar], it moves faster. It is typical for normal-phase chromatography on silica that the mobile phase is 100% organic; no water is used.
Reversed-Phase HPLC
The term reversed-phase describes the chromatography mode that is just the opposite of normal phase, namely the use of a polar mobile phase and a non-polar [hydrophobic] stationary phase. Figure S-2 illustrates the black three-dye mixture being separated using such a protocol.
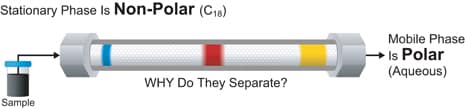
Figure S-2: Reversed-Phase Chromatography
Now the most strongly retained compound is the more non-polar blue dye, as its attraction to the non-polar stationary phase is greatest. The polar yellow dye, being weakly retained, is won in competition by the polar, aqueous mobile phase, moves the fastest through the bed, and elutes earliest like attracts like.
Today, because it is more reproducible and has broad applicability, reversed-phase chromatography is used for approximately 75% of all HPLC methods. Most of these protocols use as the mobile phase an aqueous blend of water with a miscible, polar organic solvent, such as acetonitrile or methanol. This typically ensures the proper interaction of analytes with the non-polar, hydrophobic particle surface. A C18–bonded silica [sometimes called ODS] is the most popular type of reversed-phase HPLC packing.
Table C presents a summary of the phase characteristics for the two principal HPLC separation modes based upon polarity. Remember, for these polarity-based modes, like attracts like.

Table C: Phase Characteristics for Separations Based on Polarity
Hydrophilic-Interaction Chromatography [HILIC]
HILIC may be viewed as a variant of normal-phase chromatography. In normal-phase chromatography, the mobile phase is 100% organic. Only traces of water are present in the mobile phase and in the pores of the polar packing particles. Polar analytes bind strongly to the polar stationary phase and may not elute.
Adding some water [< 20%] to the organic mobile phase [typically an aprotic solvent like acetonitrile] makes it possible to separate and elute polar compounds that are strongly retained in the normal-phase mode [or weakly retained in the reversed-phase mode]. Water, a very polar solvent, competes effectively with polar analytes for the stationary phase. HILIC may be run in either isocratic or gradient elution modes. Polar compounds that are initially attracted to the polar packing material particles can be eluted as the polarity [strength] of the mobile phase is increased [by adding more water]. Analytes are eluted in order of increasing hydrophilicity [chromatographic polarity relative to water]. Buffers or salts may be added to the mobile phase to keep ionizable analytes in a single form.
Hydrophobic-Interaction Chromatography [HIC]
HIC is a type of reversed-phase chromatography that is used to separate large biomolecules, such as proteins. It is usually desirable to maintain these molecules intact in an aqueous solution, avoiding contact with organic solvents or surfaces that might denature them. HIC takes advantage of the hydrophobic interaction of large molecules with a moderately hydrophobic stationary phase, e.g., butyl-bonded [C4], rather than octadecyl-bonded [C18], silica. Initially, higher salt concentrations in water will encourage the proteins to be retained [salted out] on the packing. Gradient separations are typically run by decreasing salt concentration. In this way, biomolecules are eluted in order of increasing hydrophobicity.
Separations Based on Charge: Ion-Exchange Chromatography [IEC]
For separations based on polarity, like is attracted to like and opposites may be repelled. In ion-exchange chromatography and other separations based upon electrical charge, the rule is reversed. Likes may repel, while opposites are attracted to each other. Stationary phases for ion-exchange separations are characterized by the nature and strength of the acidic or basic functions on their surfaces and the types of ions that they attract and retain. Cation exchange is used to retain and separate positively charged ions on a negative surface. Conversely, anion exchange is used to retain and separate negatively charged ions on a positive surface [see Figure T]. With each type of ion exchange, there are at least two general approaches for separation and elution.
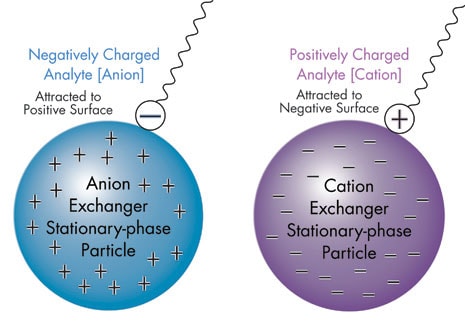
Figure T: Ion-Exchange Chromatography
Strong ion exchangers bear functional groups [e.g., quaternary amines or sulfonic acids] that are always ionized. They are typically used to retain and separate weak ions. These weak ions may be eluted by displacement with a mobile phase containing ions that are more strongly attracted to the stationary phase sites. Alternately, weak ions may be retained on the column, then neutralized by in situ changing the pH of the mobile phase, causing them to lose their attraction and elute.
Weak ion exchangers [e.g., with secondary-amine or carboxylic-acid functions] may be neutralized above or below a certain pH value and lose their ability to retain ions by charge. When charged, they are used to retain and separate strong ions. If these ions cannot be eluted by displacement, then the stationary phase exchange sites may be neutralized, shutting off the ionic attraction, and permitting elution of the charged analytes.
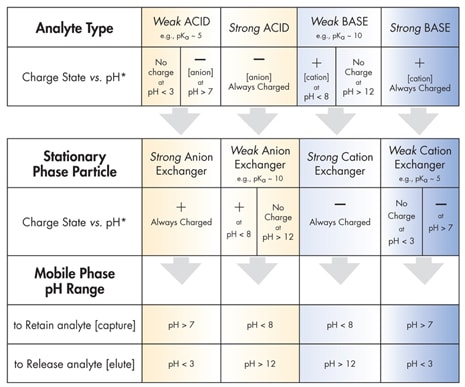
Table D: Ion-Exchange Guidelines
When weak ion exchangers are neutralized, they may retain and separate species by hydrophobic [reversed-phase] or hydrophilic [normal-phase] interactions; in these cases, elution strength is determined by the polarity of the mobile phase [Figure R-1]. Thus, weak ion exchangers may be used for mixed-mode separations [separations based on both polarity and charge].
Table D outlines guidelines for the principal categories of ion exchange. For example, to retain a strongly basic analyte [always positively charged], use a weak-cation-exchange stationary phase particle at pH > 7; this assures a negatively charged particle surface. To release or elute the strong base, lower the pH of the mobile phase below 3; this removes the surface charge and shuts off the ion-exchange retention mechanism.
Note that a pKa is the pH value at which 50% of the functional group is ionized and 50% is neutral. To assure an essentially neutral, or a fully charged, analyte or particle surface, the pH must be adjusted to a value at least 2 units beyond the pKa, as appropriate [indicated in Table D].
Do not use a strong-cation exchanger to retain a strong base; both remain charged and strongly attracted to each other, making the base nearly impossible to elute. It can only be removed by swamping the strong cation exchanger with a competing base that exhibits even stronger retention and displaces the compound of interest by winning the competition for the active exchange sites. This approach is rarely practical, or safe, in HPLC and SPE. [Very strong acids and bases are dangerous to work with, and they may be corrosive to materials of construction used in HPLC fluidics!]
Separations Based on Size: Size-Exclusion Chromatography [SEC] –
Gel-Permeation Chromatography [GPC]
In the 1950s, Porath and Flodin discovered that biomolecules could be separated based on their size, rather than on their charge or polarity, by passing, or filtering, them through a controlled-porosity, hydrophilic dextran polymer. This process was termed gel filtration. Later, an analogous scheme was used to separate synthetic oligomers and polymers using organic-polymer packings with specific pore-size ranges. This process was called gel-permeation chromatography [GPC]. Similar separations done using controlled-porosity silica packings were called size-exclusion chromatography [SEC]. Introduced in 1963, the first commercial HPLC instruments were designed for GPC applications [see Reference 3].
All of these techniques are typically done on stationary phases that have been synthesized with a pore-size distribution over a range that permits the analytes of interest to enter, or to be excluded from, more or less of the pore volume of the packing. Smaller molecules penetrate more of the pores on their passage through the bed. Larger molecules may only penetrate pores above a certain size so they spend less time in the bed. The biggest molecules may be totally excluded from pores and pass only between the particles, eluting very quickly in a small volume. Mobile phases are chosen for two reasons: first, they are good solvents for the analytes; and, second, they may prevent any interactions [based on polarity or charge] between the analytes and the stationary phase surface. In this way, the larger molecules elute first, while the smaller molecules travel slower [because they move into and out of more of the pores] and elute later, in decreasing order of their size in solution. Hence the simple rule: Big ones come out first.
Since it is possible to correlate the molecular weight of a polymer with its size in solution, GPC revolutionized measurement of the molecular-weight distribution of polymers that, in turn, determines the physical characteristics that may enhance, or detract from, polymer processing, quality, and performance [how to tell good from bad polymer].
Conclusion
We hope you have enjoyed this brief introduction to HPLC. We encourage you to read the references below and to study the Appendix on HPLC Nomenclature.