Quantitation and Calibration
When a compound is already known, as in the case of clinical trials where statistical data from many individual samples is gathered and the administered drug and its metabolite of interest are already well characterized, full mass spectra are not required. However, very good sensitivity in a complex physiological mixture is required, so the instrument is set to monitor only specific m/z values. (Refer to the comparison of SIR and MRM response, previously, in this primer).
Because ions continuously stream through the triple or tandem quadrupole there is no need to limit the ion current into the mass analyzer. The ion trap, on the other hand, has a defined, finite volume and therefore needs a function to prevent too many ions entering the trap. Not controlling ion intensity results in undesirable or unexpected peaks in a spectrum, a particularly troublesome phenomenon when attempting to library search EI spectra in GC/MS. The design of ion traps has changed substantially to allow external (ions created outside the mass filter with subsequent ion injection into the trap) rather than internal ionization (ionization within the mass filter). The design change addressed the problem of ion molecule reactions in the trap, but it also introduced a limitation. Even in MRM mode this automatic governing of the total ion current can result in irregular sampling intervals across a chromatographic peak. So, ultimately, the ion trap is limited as a tool for trace analysis in complex matrices, particularly when the highest accuracy and precision is required, as is the case, for data which must be legally defensible or whose stringent criteria for accuracy and precision of quantitation is dictated by legislation.
An internal standard is typically used when performing MS quantitation. The standard provides controls over variability in extraction processes, LC injection, and ionization. Without an internal standard RSDs among replicates can be tenfold higher than replicates referenced to a standard that typically produces RSDs in low single digits. The best internal standard is an isotopically labeled version of the molecule of interest. Although synthesizing such a molecule prove expensive, it will exhibit similar extraction recovery, chromatographic retention time, and ionization response in the mass spectrometer.
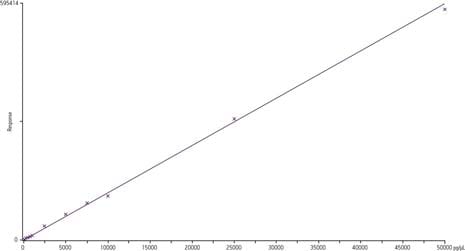
Quantitation example using GC/MS with CI - Linear Dynamic Range 5 Orders
Assessing system suitability, randomizing samples, and determining suitable curves and concentration points are the subject of lengthy debate. Some good references can be seen at the https://www.ionsource.com/tutorial/msquan/requantoc.htm.
Calibration
Calibration compounds are used by a mass spectrometrist to adjust the mass calibration scale, as well as the relative intensities of the ions, to match that of known entities. This operation is performed on all mass spectrometers because subtle changes in electronics, cleanliness of surfaces, and a lab’s ambient conditions can affect the instrument’s ability to reproduce a meaningful measurement. For the least demanding analyses on nominal-mass instruments, the need for calibration can be infrequent and a check of its response more frequent. Nevertheless, high mass accuracy requires constant surveillance for minute changes.
For GCMS, a popular calibration compound is FC-43, also known as perfluorotributylamine. Other calibration compound mixtures are used to adjust a high-resolution mass spectrometer’s calibration scale. Sodium cesium iodide (NaCsI) and polyethylene glycol mixtures are popular for LCMS. In an LCMS-compatible solvent, NaCsI streamed or infused in a steady state into an instrument provides a series of monoisotopic peaks through 4000 Da.
Kits are available that contain a selection of standard peptides, proteins, matrices, and solvents for calibrating, tuning, and sensitivity testing matrix-assisted, laser-desorption ionization (MALDI) mass spectrometers. One, from Sigma Aldrich, is a setup aid for analyzing complex mixtures of proteins and peptides (700 to 66,000 Da).
Lock mass
Constant vigilance is required for the most demanding measurement with TOFs and similar highly accurate instruments. A small change in temperature alone can shift the reported mass results by many parts per million. Depending on the type of ionization used constant calibration correction can be achieved by simply using a known contaminant present in the source. Or you can sample an ion stream periodically to reestablish proper calibration throughout an analysis. Simply adding a lock mass calibrant to flowing LC eluent, by “teeing in”’ after the column and before the mass spectrometer’s inlet, often causes uncontrolled behaviors like ion suppression, mass interference, and solvent effects.
Time-of-flight (TOF) instruments (described earlier in this primer) can reach low parts per million accuracy, and Fourier transform ion cyclotron resonance (FTICR) instruments are capable of even greater accuracy, provided the number of ions admitted are well controlled. Real-time recalibration on the lock mass by corrections of any mass shift removes mass error relative to that established with calibration of the mass scale. Weak signal is another sometimes-overlooked cause, which is correctable by averaging the mass measurement over the LC peak, weighted by signal intensity.
A dual-electrospray source optimized for acquiring exact mass data is ideal for proteomics studies or low-level metabolite identification. The method using two independent ESI probes samples the correcting spray (or reference) stream, using an oscillating baffle driven by a programmable stepper motor. The reference spray is sampled at predefined intervals, ensuring the acquisition duty cycle favors the liquid stream containing the analyte. The sampling baffle position is monitored in real-time, enabling indexing of the two liquid inlets, and the reference and sample data are stored in separate files. The design eliminates cross-talk between the analyte and reference channels.