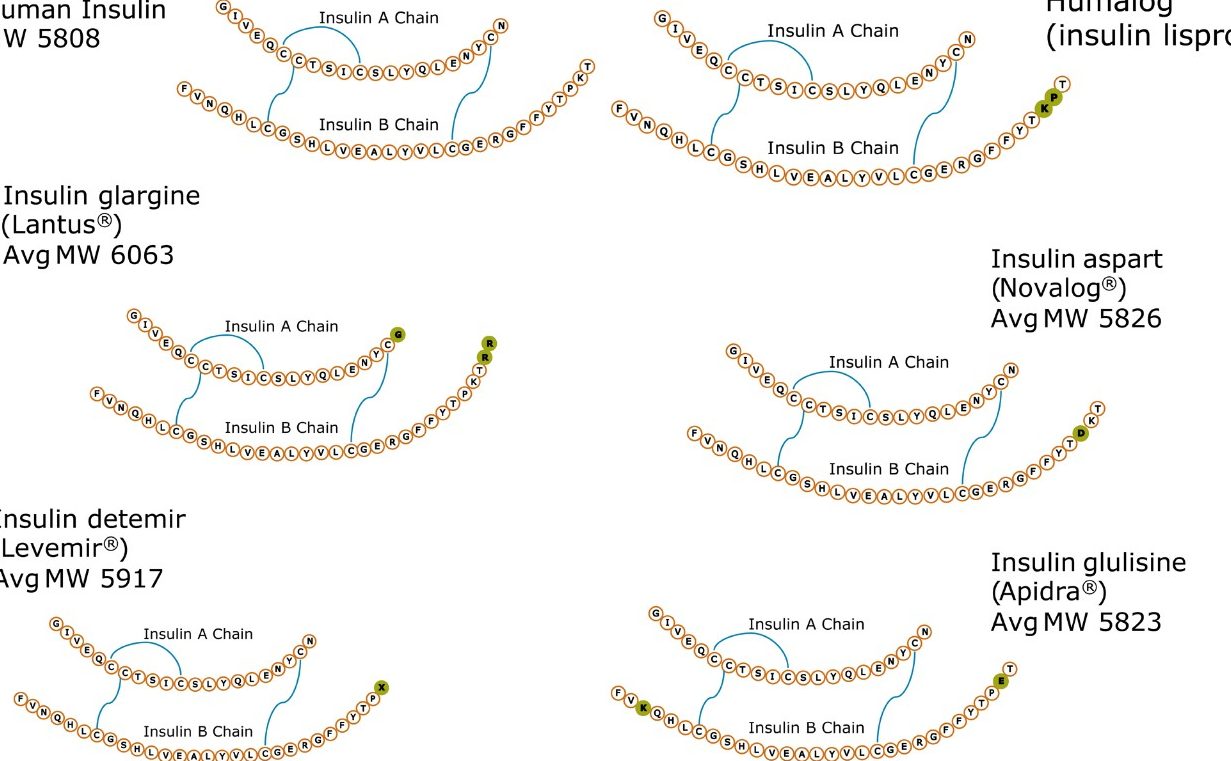
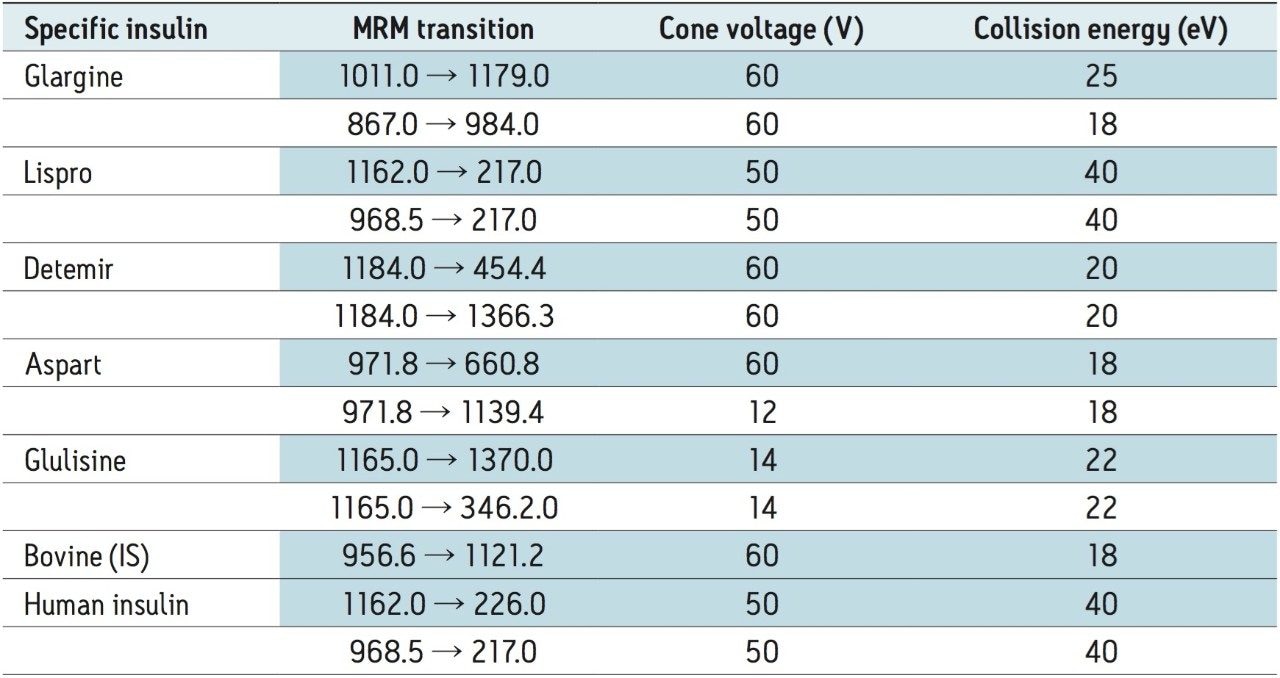
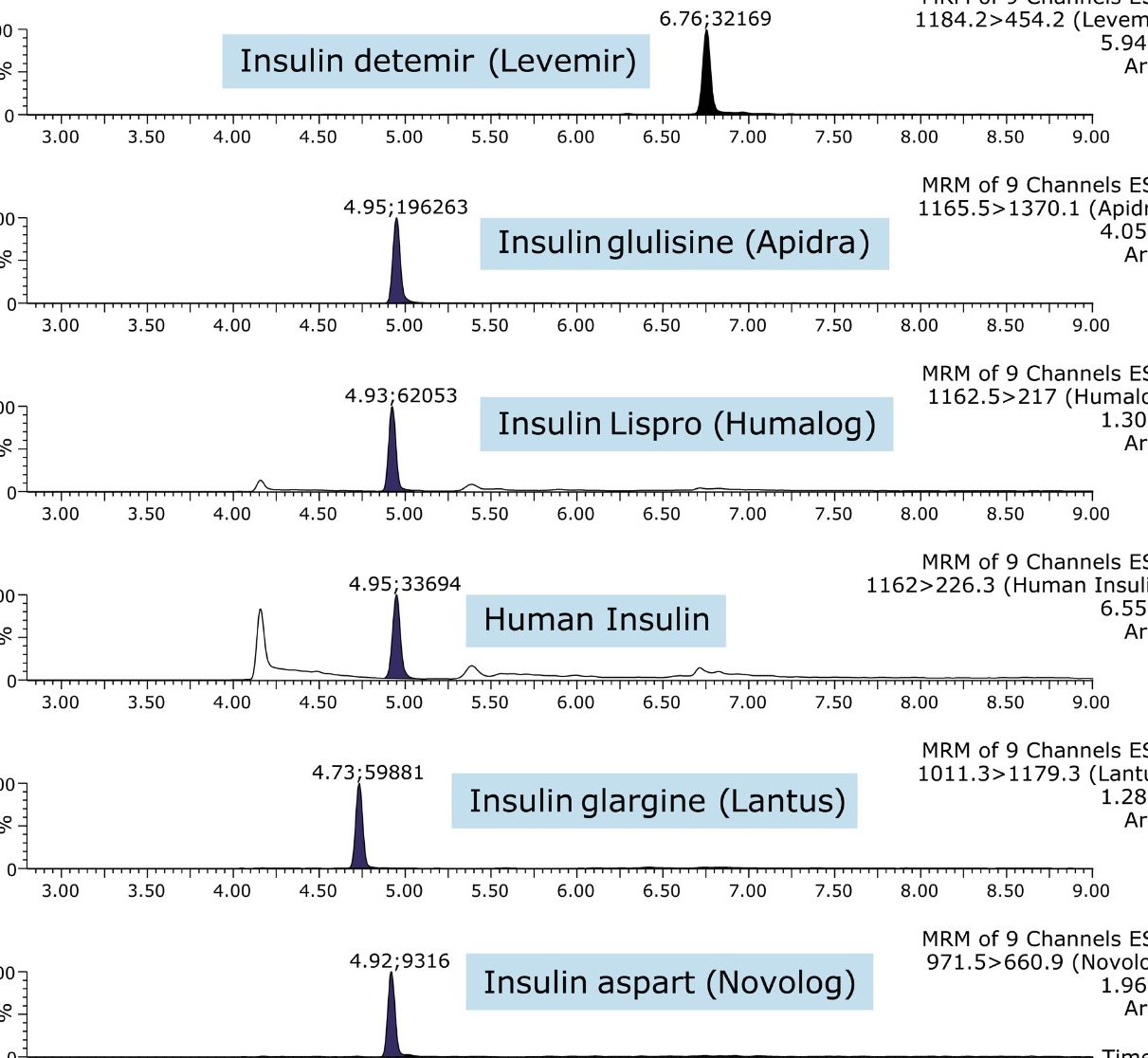
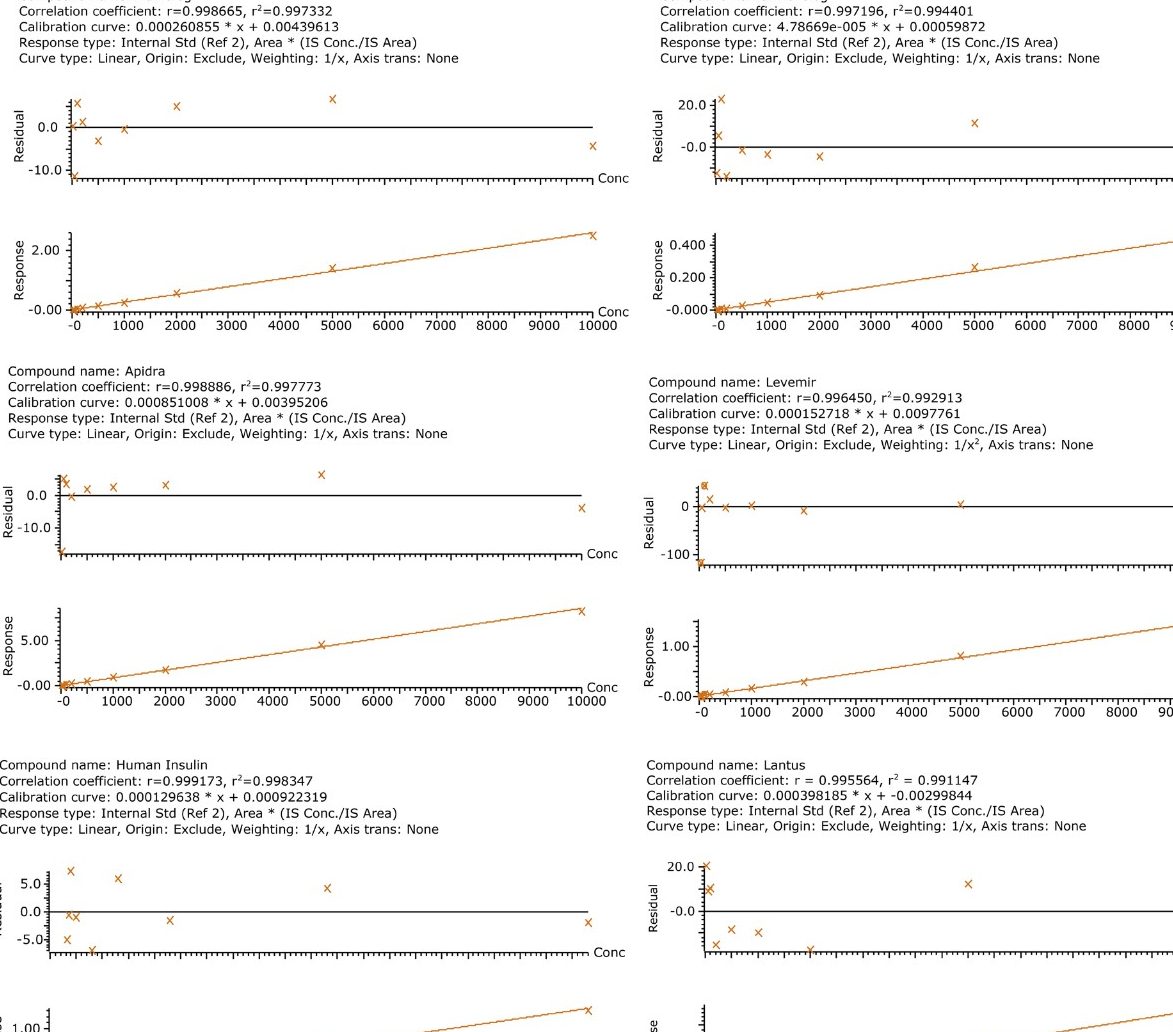
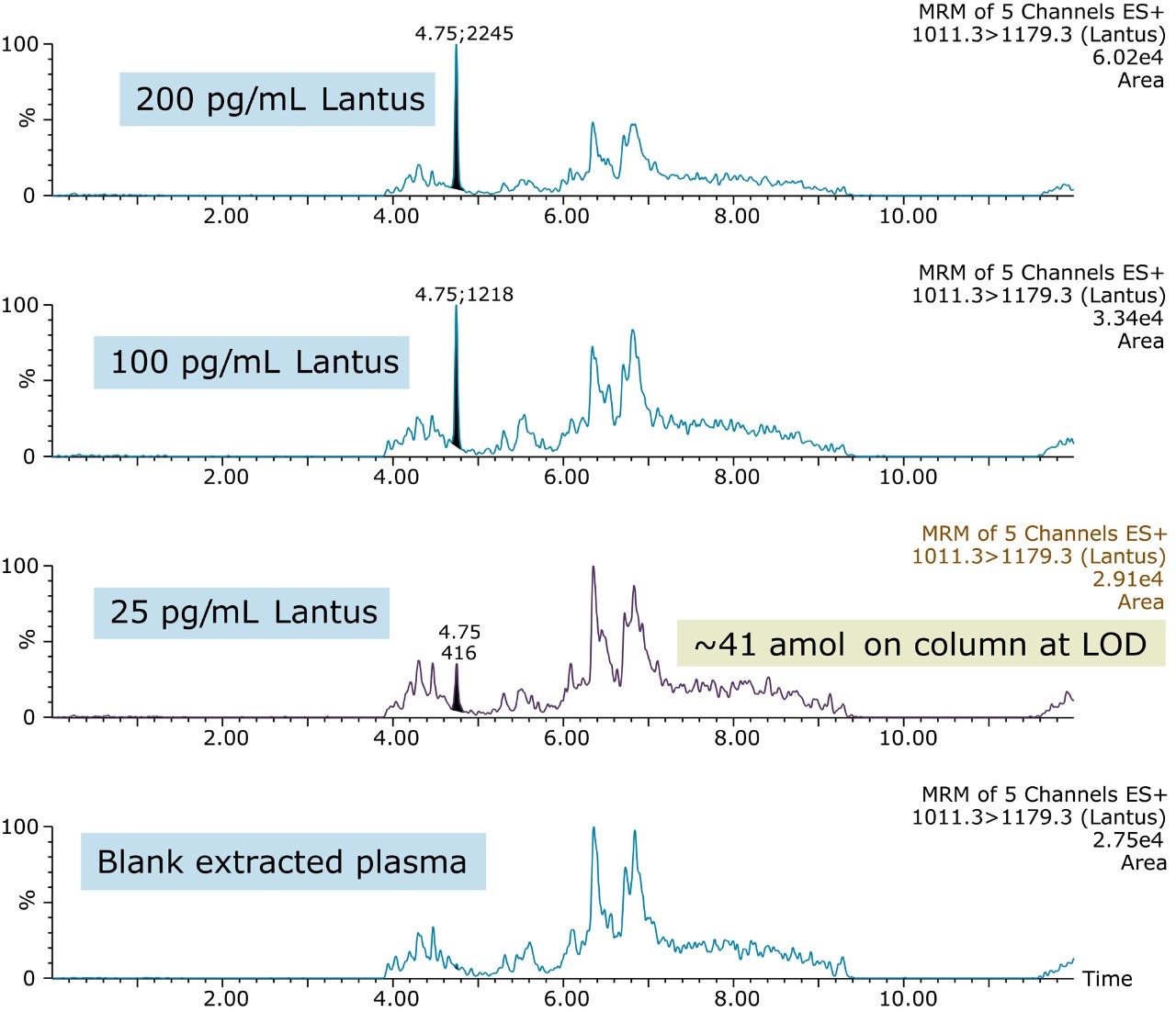
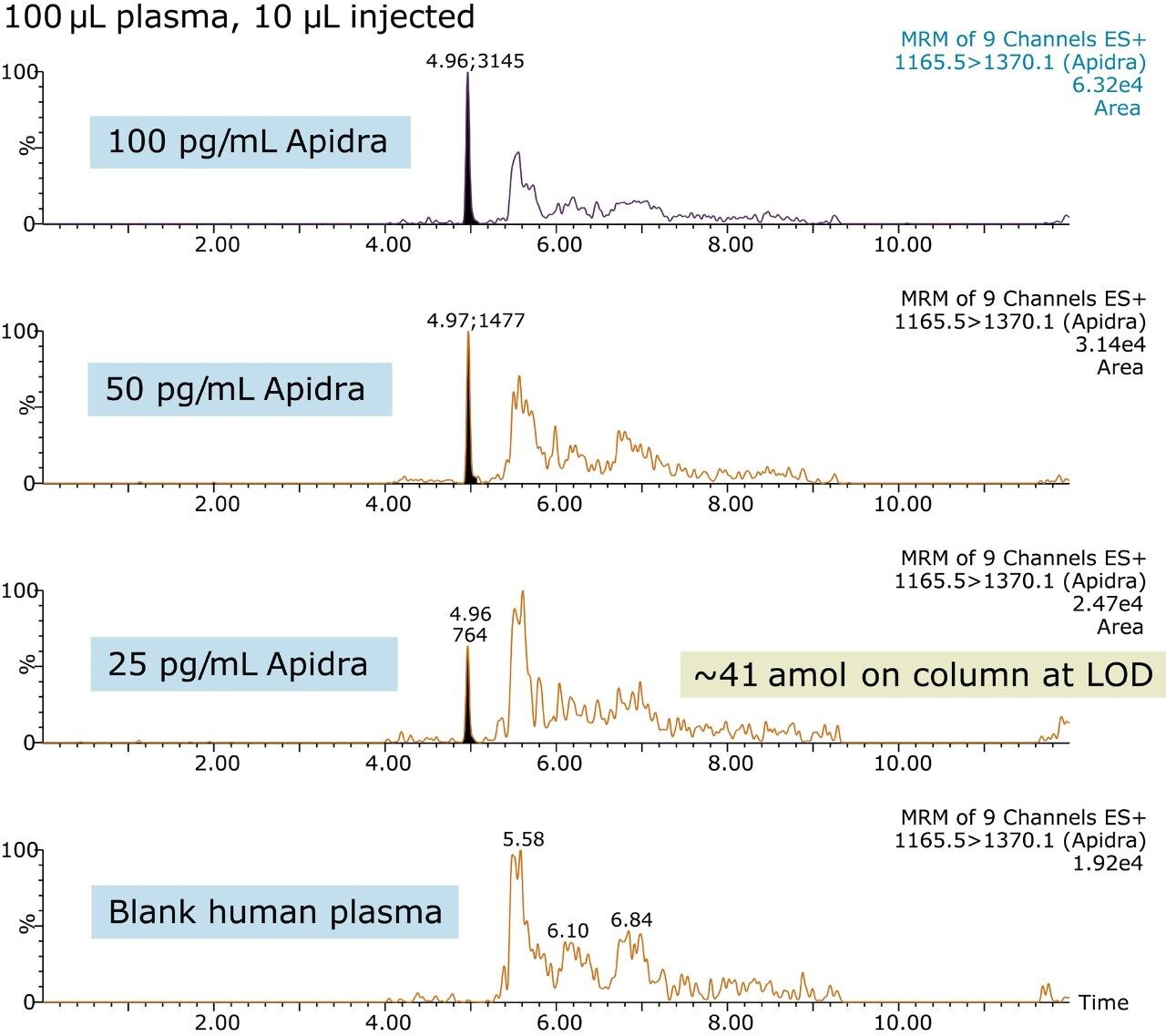
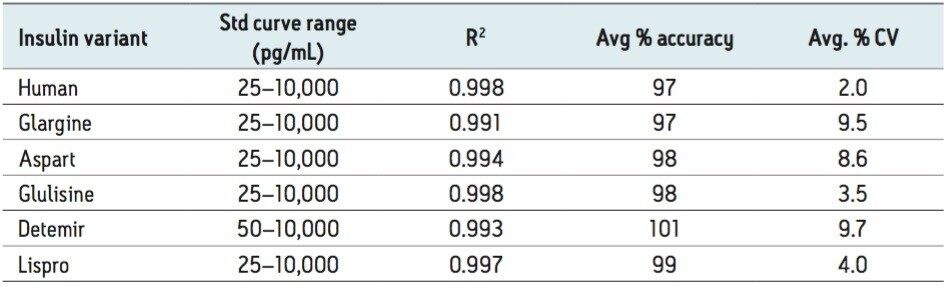
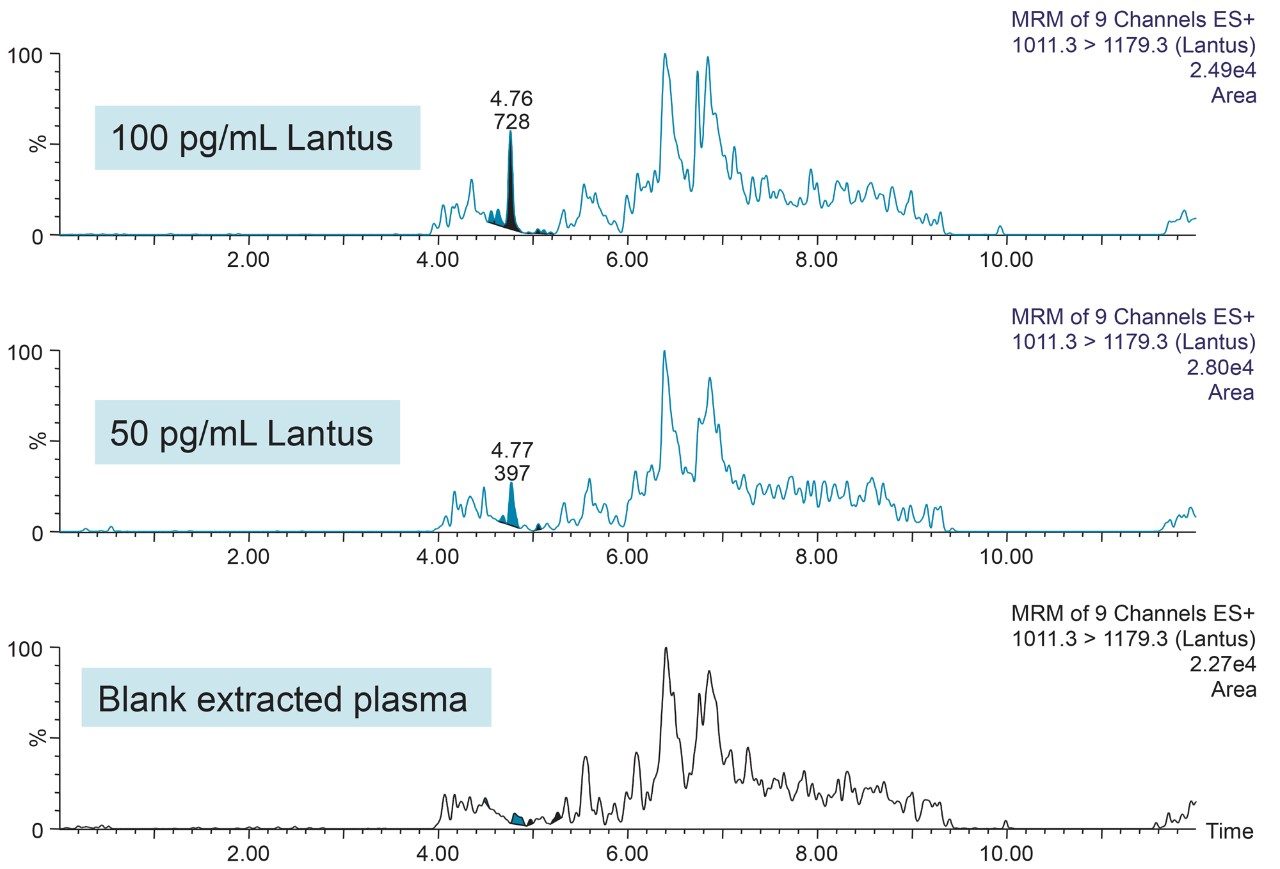
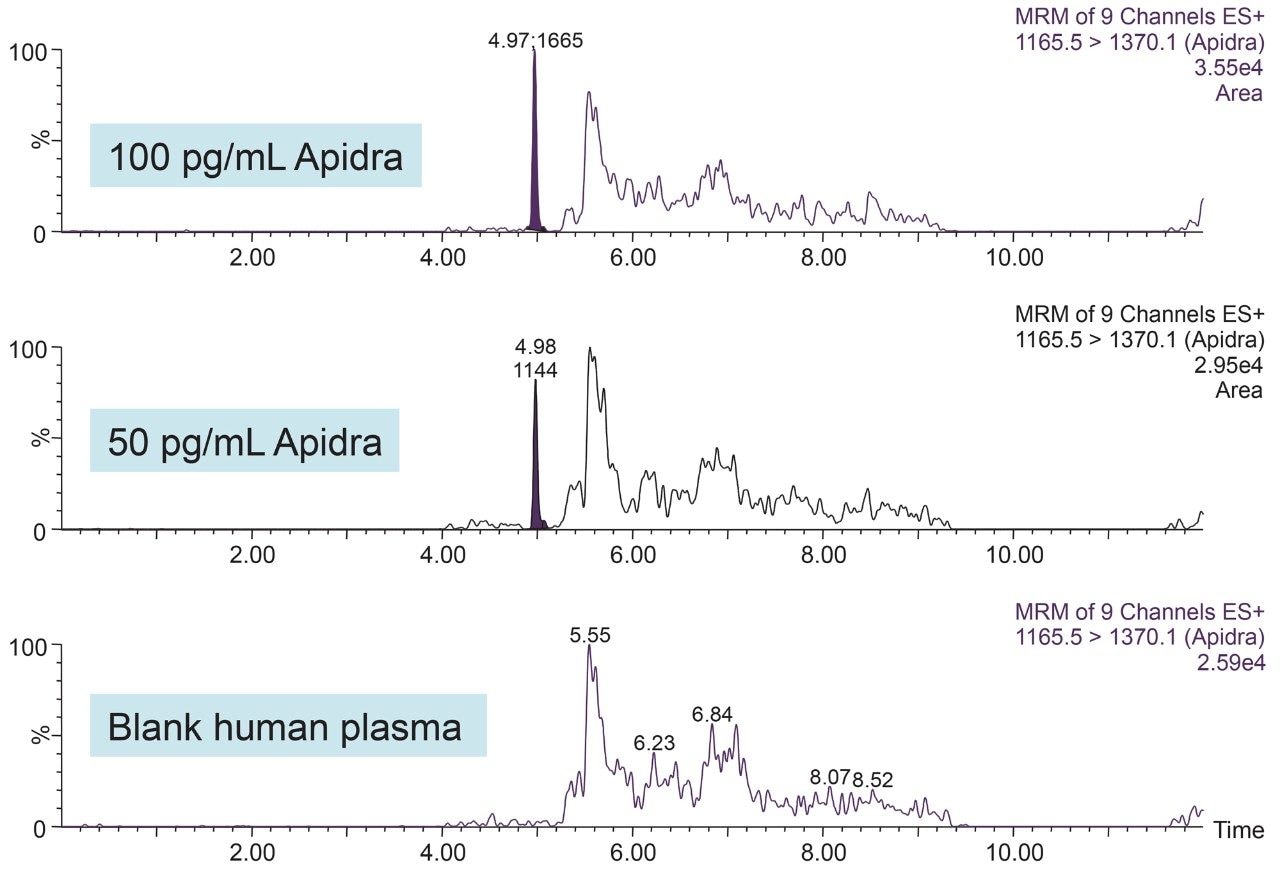
In an earlier publication,1 we described an ultra-high sensitivity quantitative assay for human insulin and 5 analogs. The method was carefully optimized to maximize sensitivity for the insulins in the following manner: a multi-dimensional LC system was used to enable at-column-dilution and a trap/back elute strategy to increase loading volume and then refocus the analyte band. Mixed-mode SPE and a high-efficiency chromatographic system using a solid-core column with a positively-charged particle surface improved specificity and facilitated the differentiation of human insulin and insulin LisPro. In this earlier method, 250 µL of human plasma were extracted to reach detection limits between 50 and 200 pg/mL with a 30 µL injection volume.
Many of the insulins described in the earlier method have either recently come off patent or are due to shortly. This has resulted in a flurry of research activity aimed at alternate dosing regimes, pediatric extensions, and the development of replacement insulins. In many of these cases, a further decrease in detection limit and reduction in sample volume required were requested.
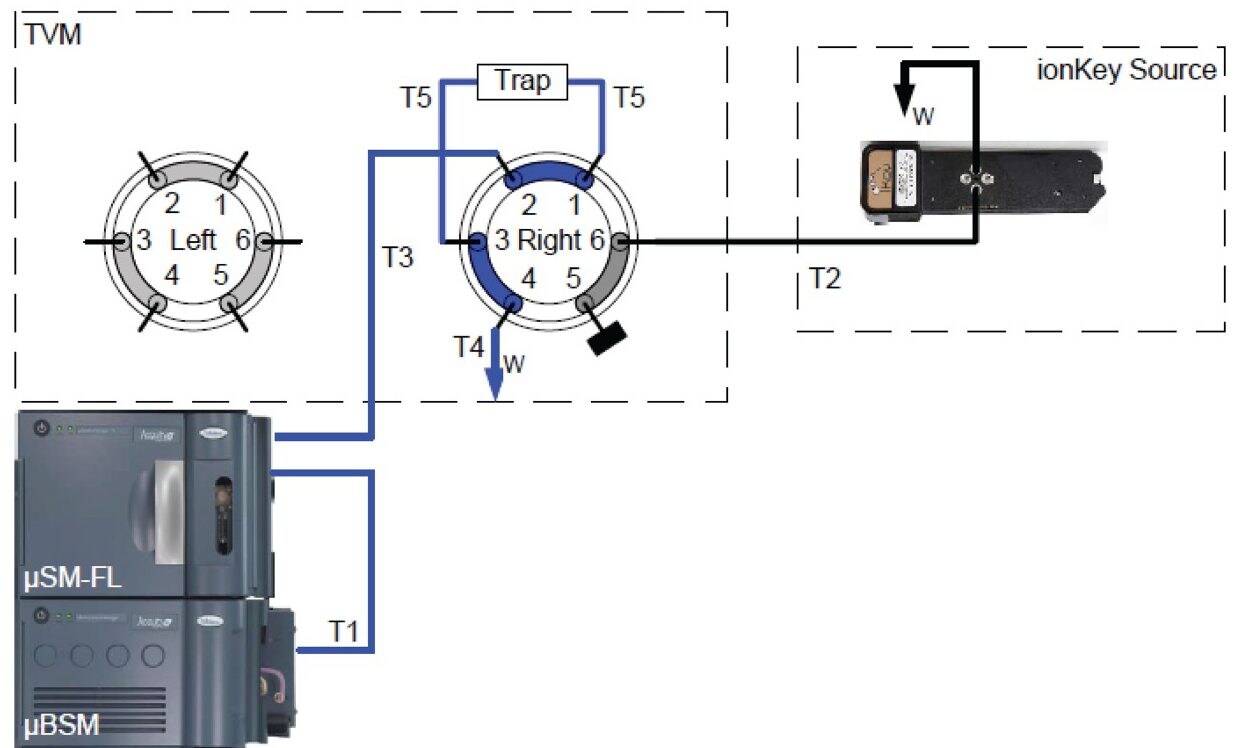
In this current work, we undertook to a) transfer the original analytical scale method to the ionKey/MS System, and b) to decrease sample volume and further increase sensitivity through the inherent characteristics of the ionKey/MS System. The ionKey/MS System integrates UPLC analytical separation directly into the source of the mass spectrometer. The iKey Separation Device (150 µm internal diameter), shown in Figure 2, contains the fluidic channel, electronics, ESI interface, heater, eCord, and the chemistry to perform UPLC separations. Perhaps most importantly, The ionKey/MS System can provide increased sensitivity compared to 2.1 mm chromatography with the same injection volume, or equivalent or greater sensitivity with reduced sample consumption, making it ideal for insulin analyses. As previously mentioned, it is common for bioanalytical LC-MS assays to consume high volumes of both solvent and sample, thus increasing the cost of the assay and limiting the number of replicates that can be analyzed. This study combines µElution solid-phase extraction (SPE) and the novel and highly efficient ionKey/MS System to improve a quantitative assay for insulins in human plasma.