Ranitidine is a histamine-2 blocker, which decreases the amount of acid created by the stomach and is approved for multiple indications, including treatment and prevention of stomach and intestinal ulcers, as well as treatment of gastroesophageal reflux disease.1 Ranitidine is manufactured by many pharmaceutical and generic companies and is available over the counter (OTC) and by prescription. In 2019, reports appeared that the N-nitrosamine impurity, N-nitrosdimethylamine (NDMA) was found to be present in ranitidine drug products and resulted in recalls of this product.2,3
N-nitrosamines, as a class, are known environmental contaminants with suspected carcinogenic/genotoxic effects in animals and humans.4,5 In response to public concern, regulatory agencies have issued guidance for allowable limits of these genotoxic impurities (GTIs) with an acceptable daily intake limit of 96 ng/day (0.32 ppm) for NDMA in ranitidine and a proposed limit in the future of 0.03 ppm. Information on how to assess and control these impurities can be found in the ICH M7 (R1) guideline.7
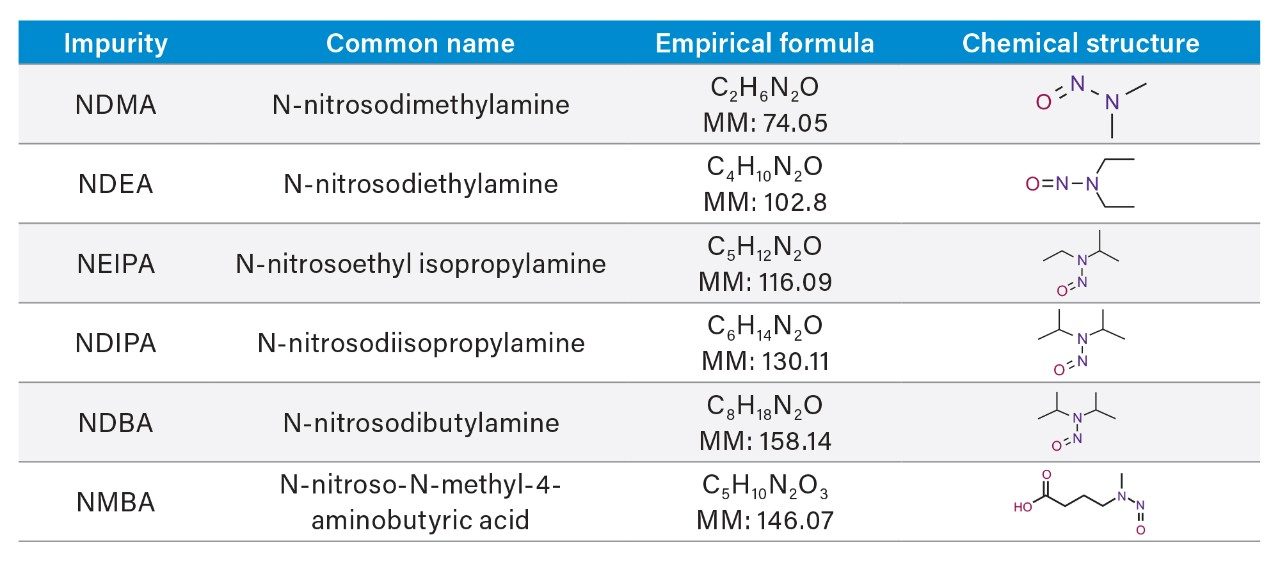
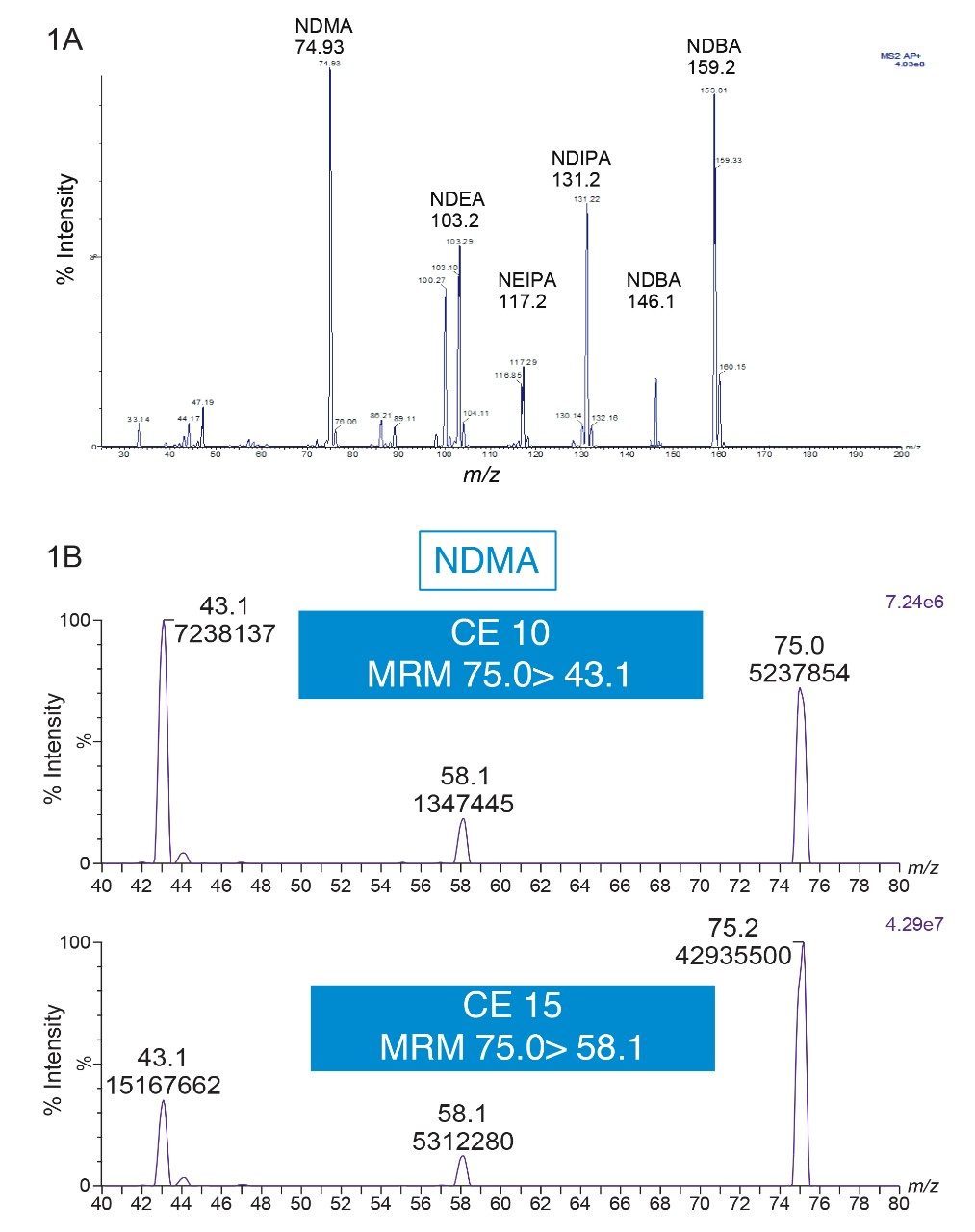
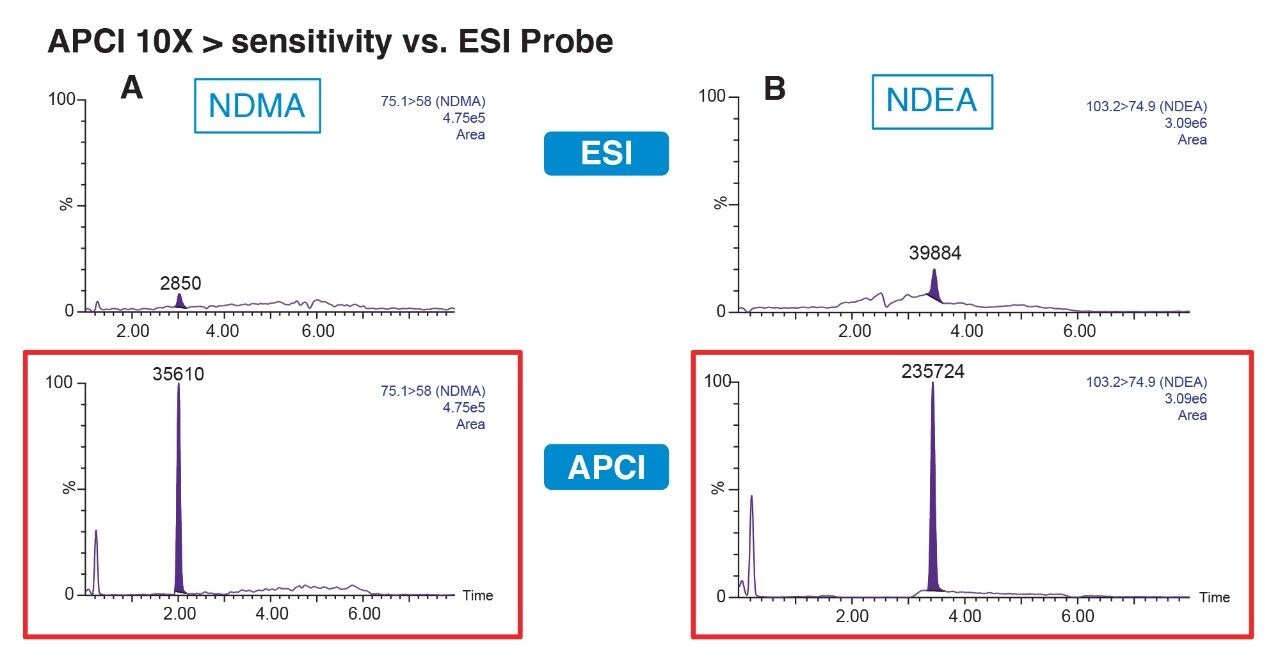
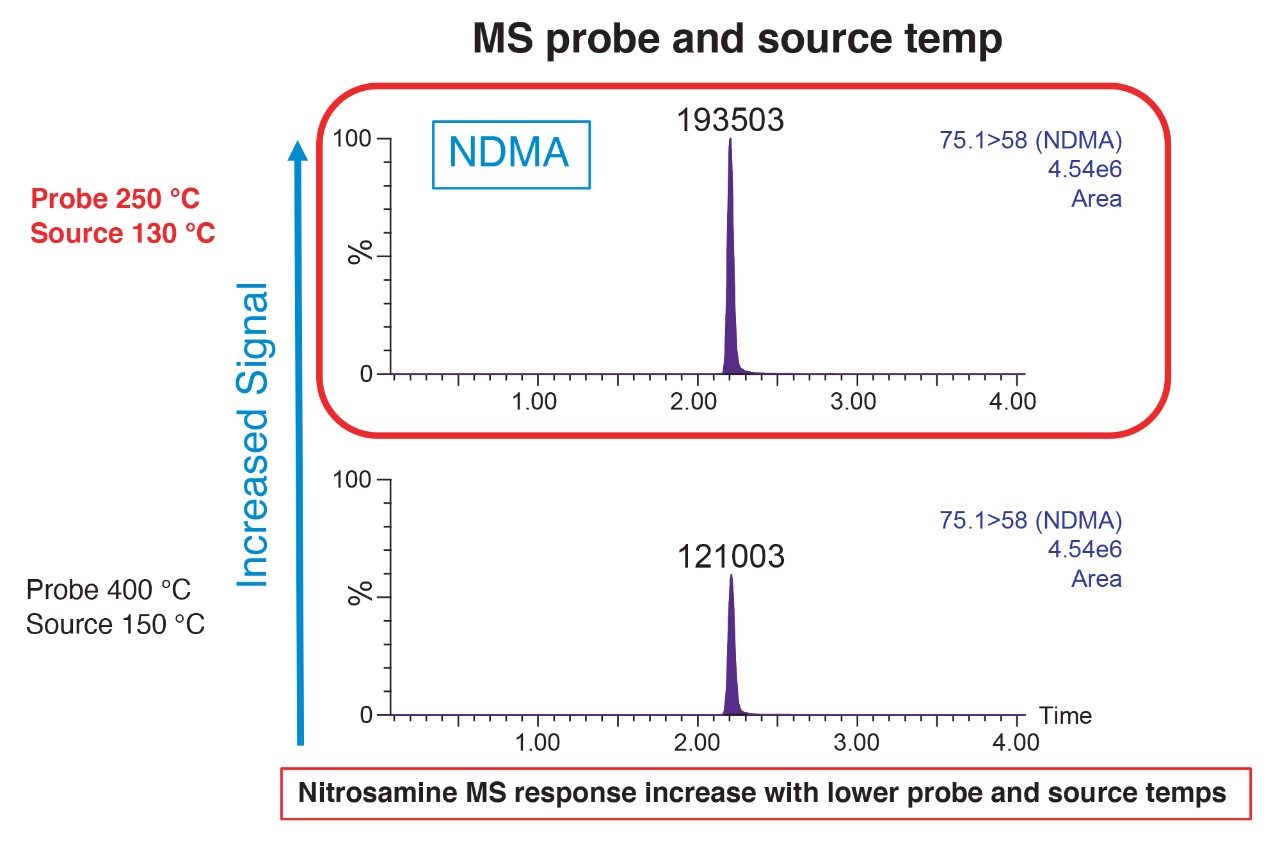
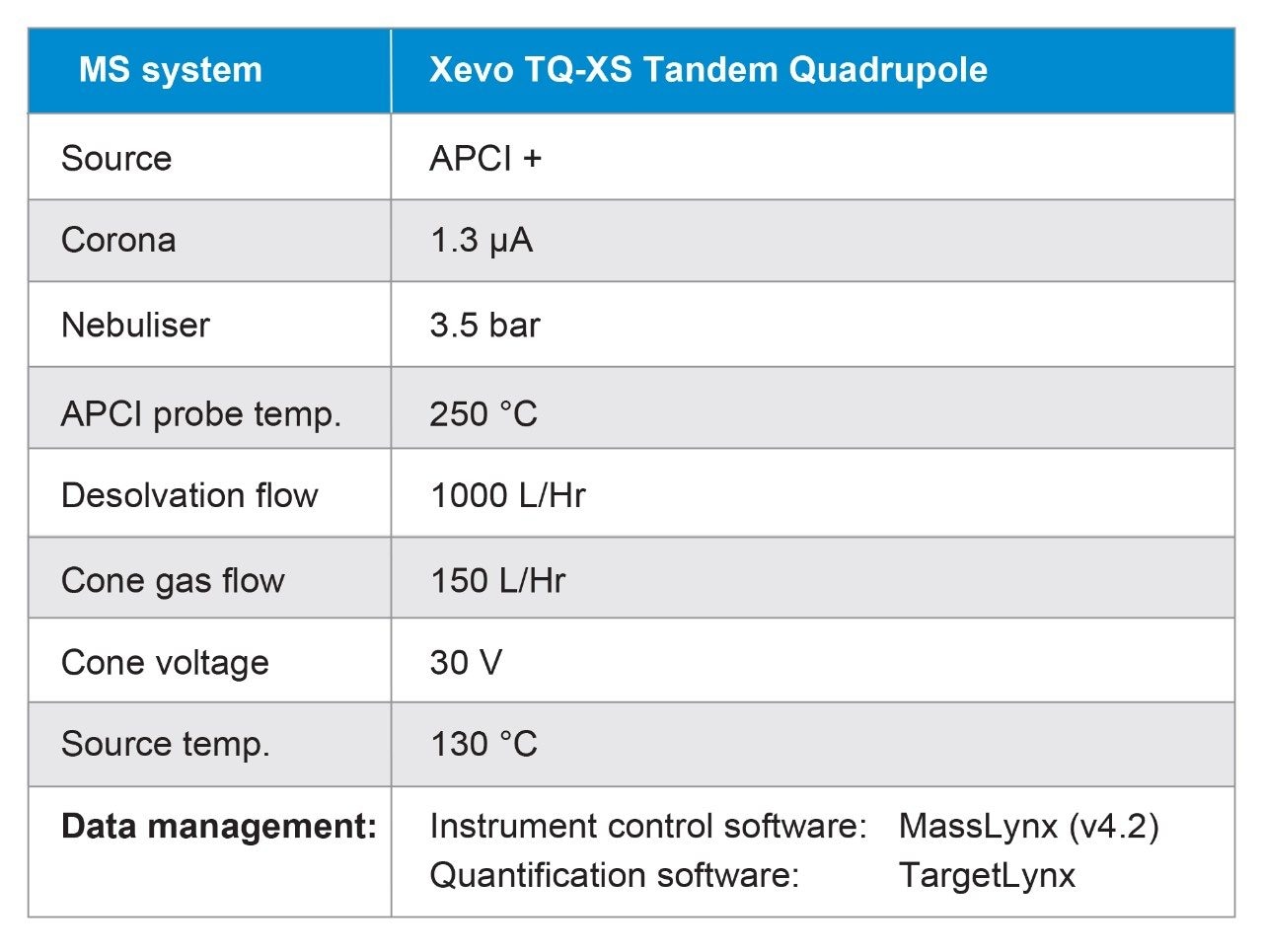
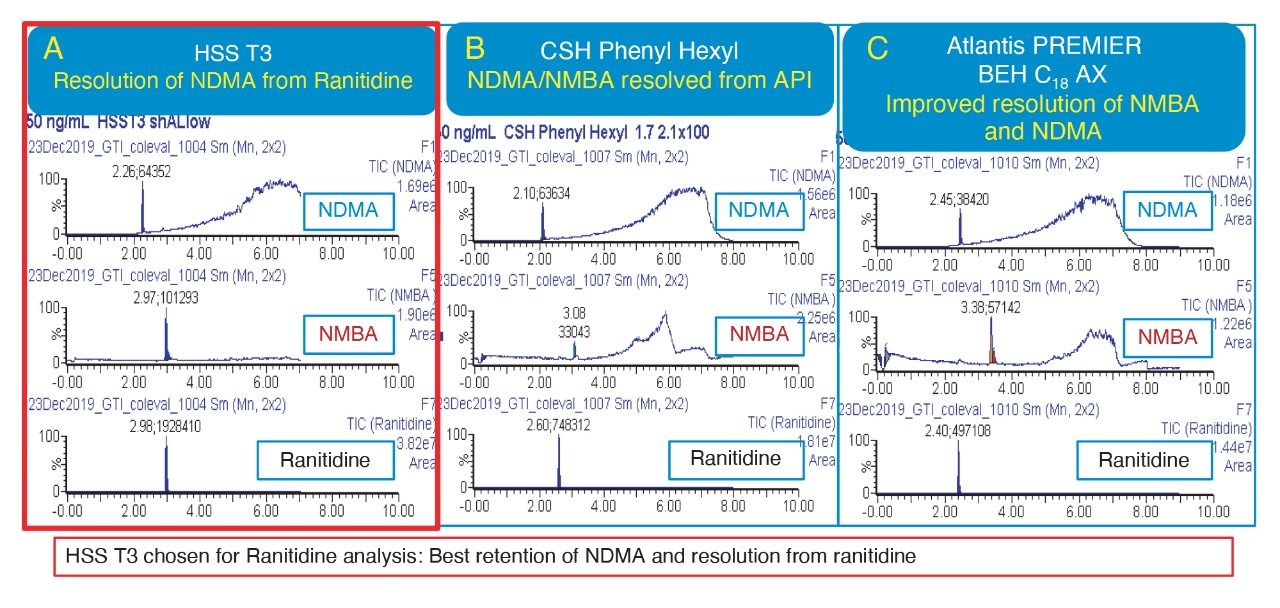
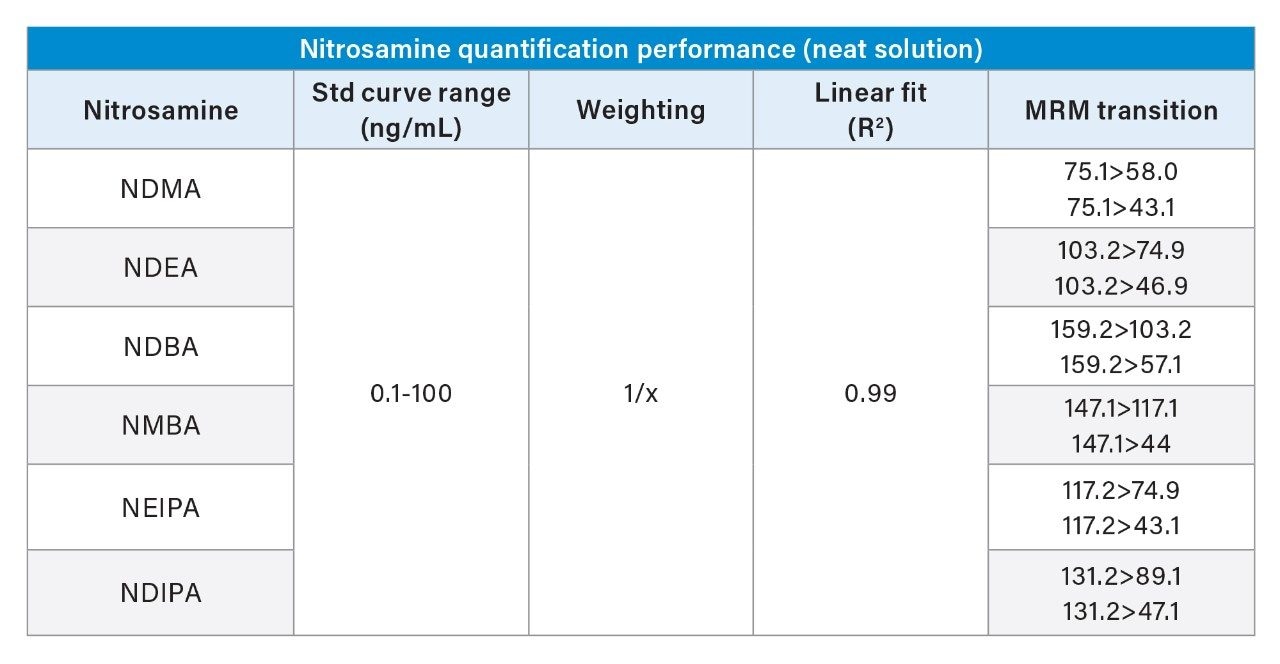
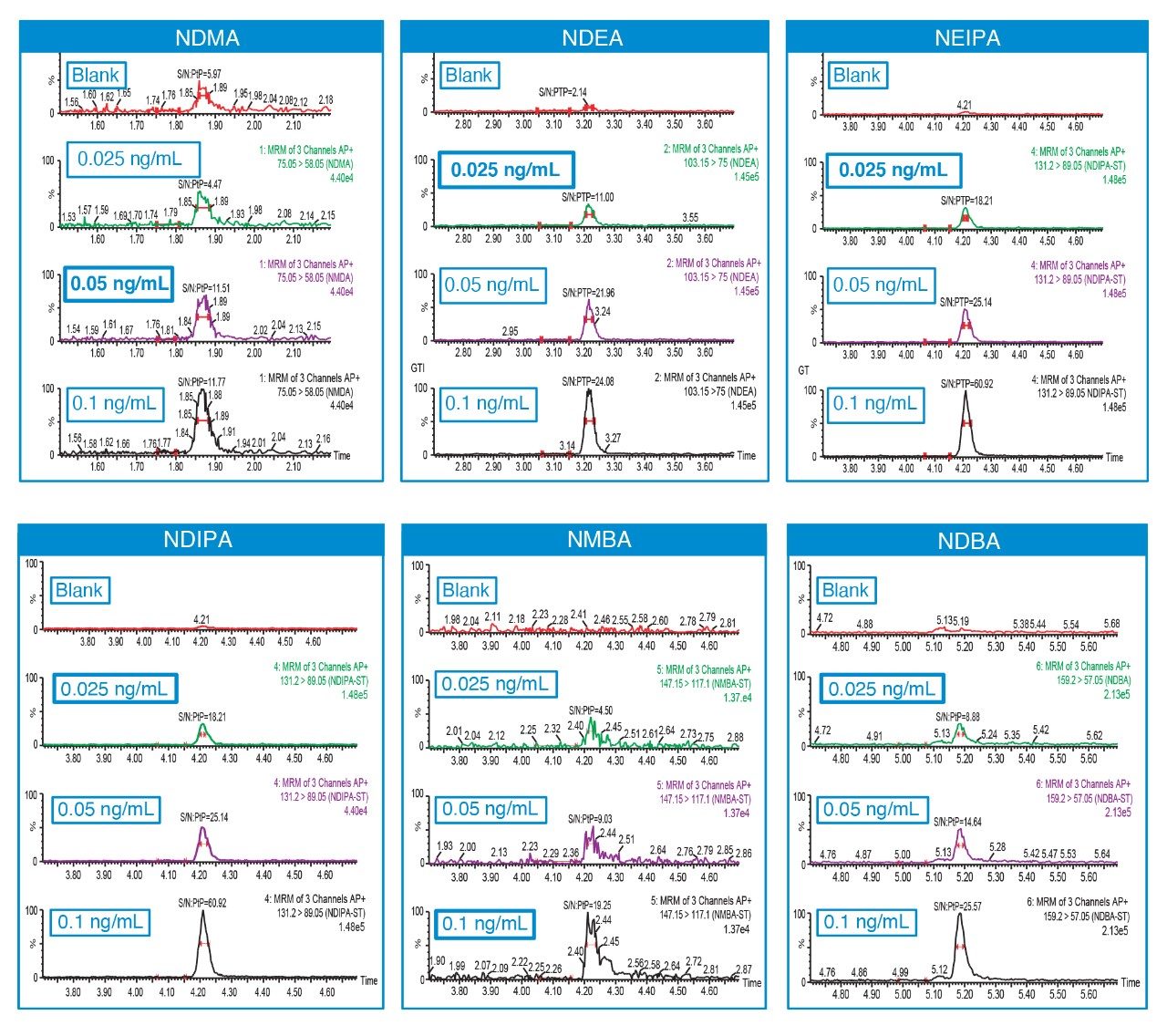
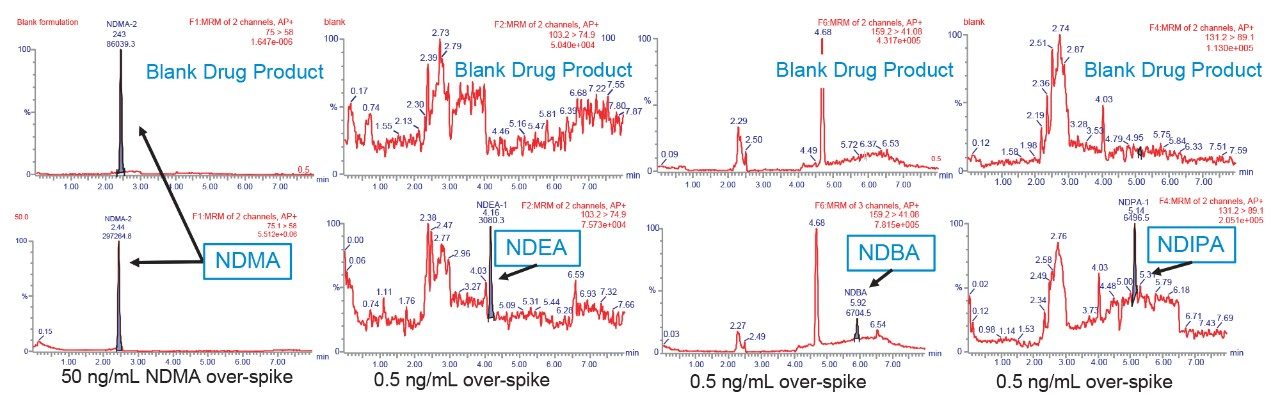
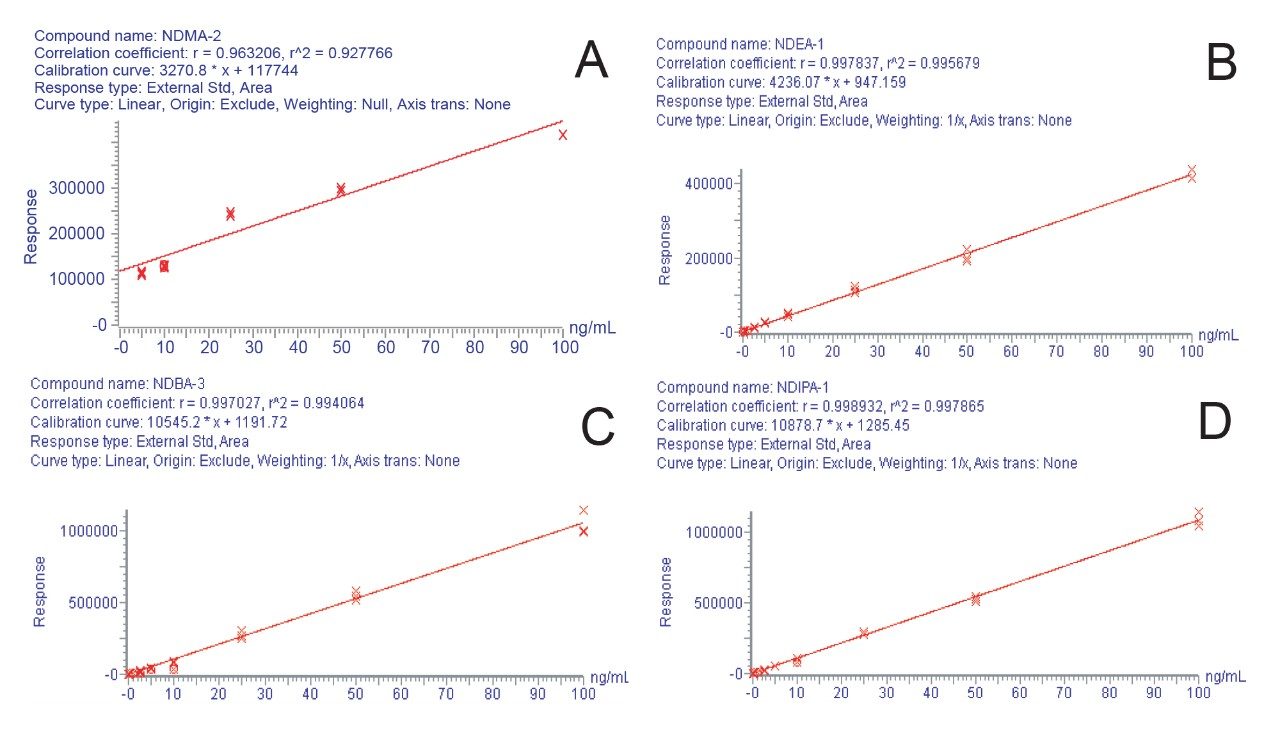
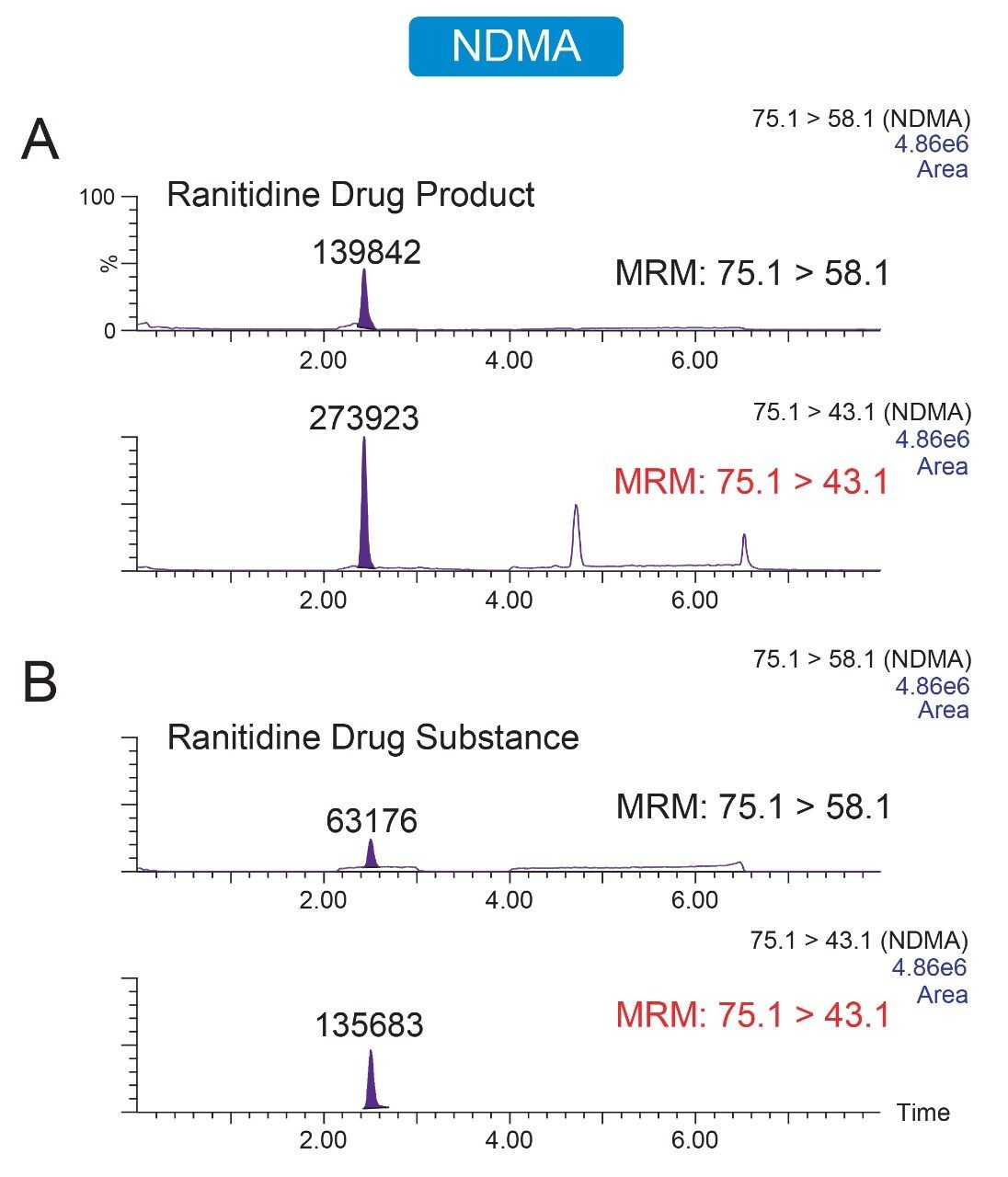
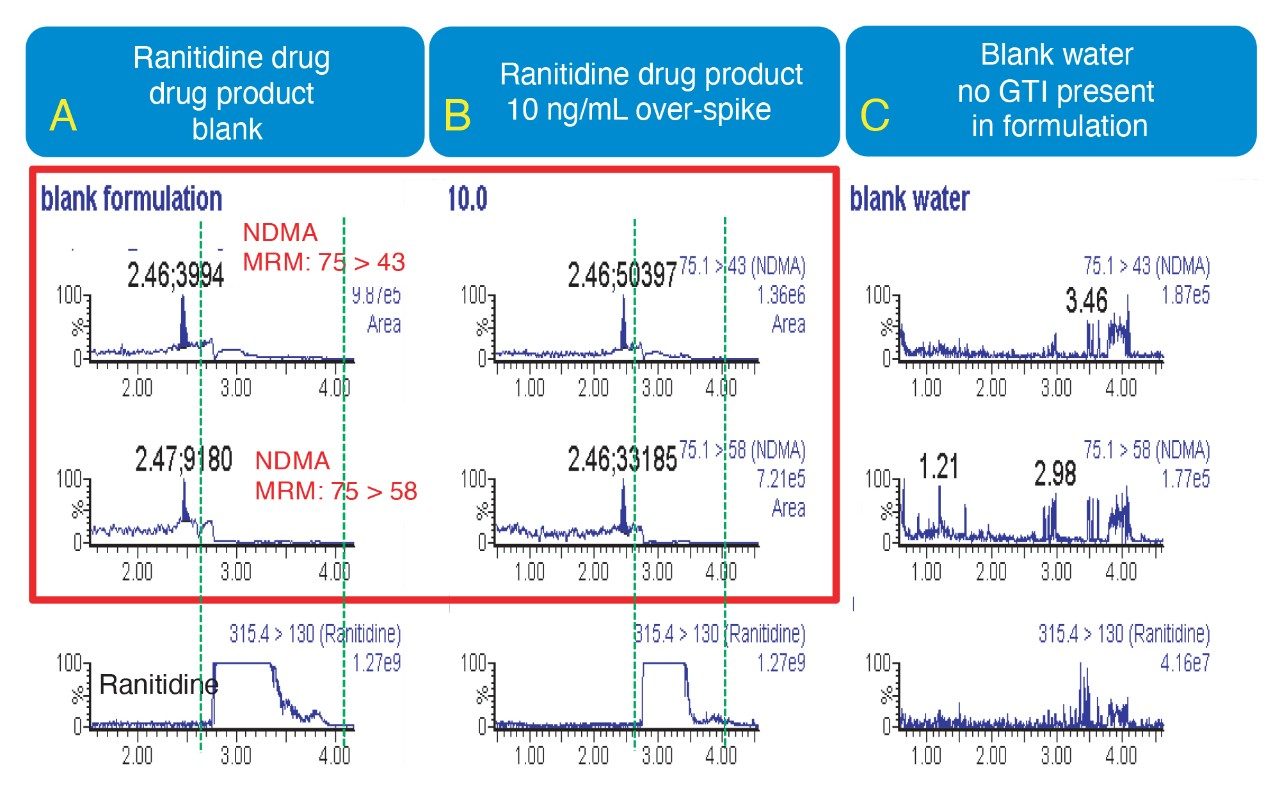
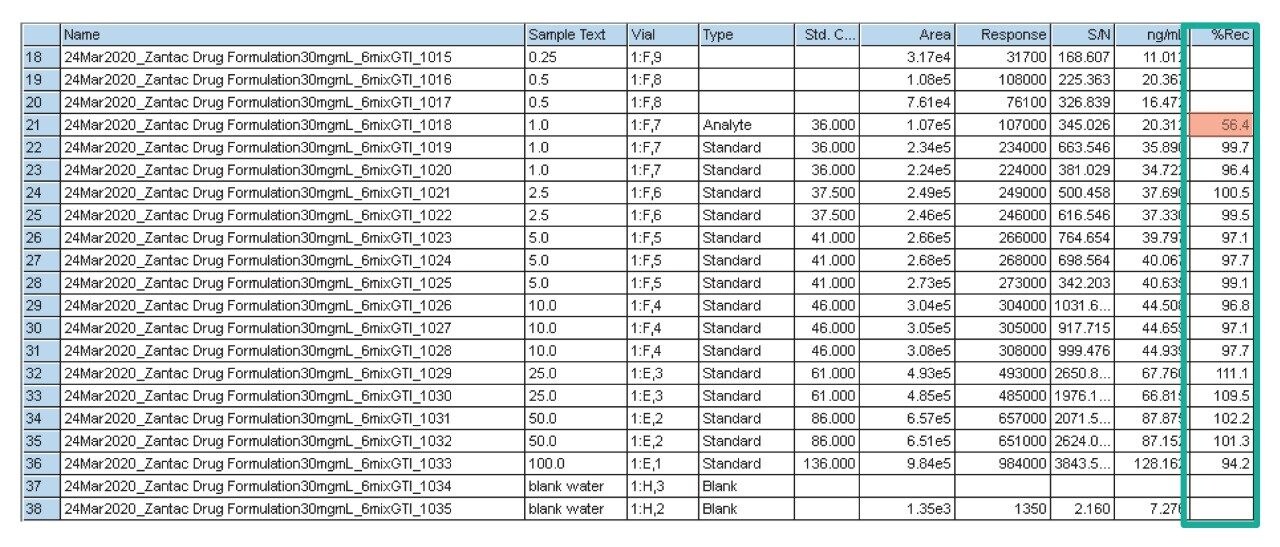
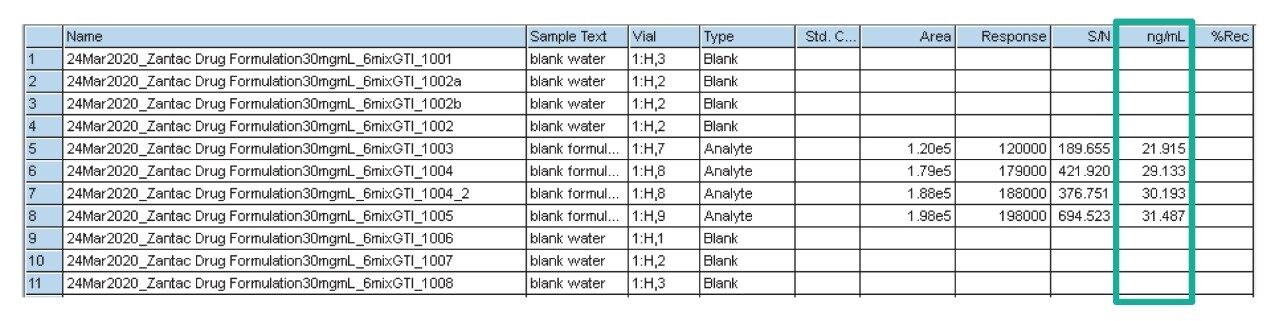
Due to the regulatory guidance’s low safety threshold levels for these compounds, there exists a strong need for LC-MS methods that can accurately quantify them at low ppm levels. Developing such methods is challenging due to the chemical diversity of nitrosamines, poor chromatographic retention, MS ionization, and fragmentation, often limiting sensitivity and selectivity. This work presented herein, provides practical considerations for optimization of LC-MS conditions to achieve sensitive and robust simultaneous quantification of several nitrosamine GTIs (NDMA, NDEA, NEIPA, NDIPA, NDBA, and NMBA). A list of these impurities, including their chemical information, is shown in Table 1. The developed analytical method employs ultra performance liquid chromatography (UPLC) and tandem quadrupole MS-MS detection. Using the low dispersion ACQUITY UPLC I-Class PLUS and reversed-phase (UPLC-RP) separation with a sub-2-μm C18 column designed specifically for retention and separation of polar compounds coupled to a high sensitivity tandem quadrupole MS, lower limits of quantification (LLOQ) between 0.025–0.1 ng/mL (<1 pg on column) in ranitidine drug substance and product were achieved. This method was used to analyze a ranitidine drug product tablet, achieving an LLOQ of 0.1 ng/mL (0.0025 ppm based on a 30 mg/mL dose) and determining the concentration of NDMA in the tablet to be 29.0 ng/mL, or 1 ppm relative to the ranitidine API.