Control Strategy
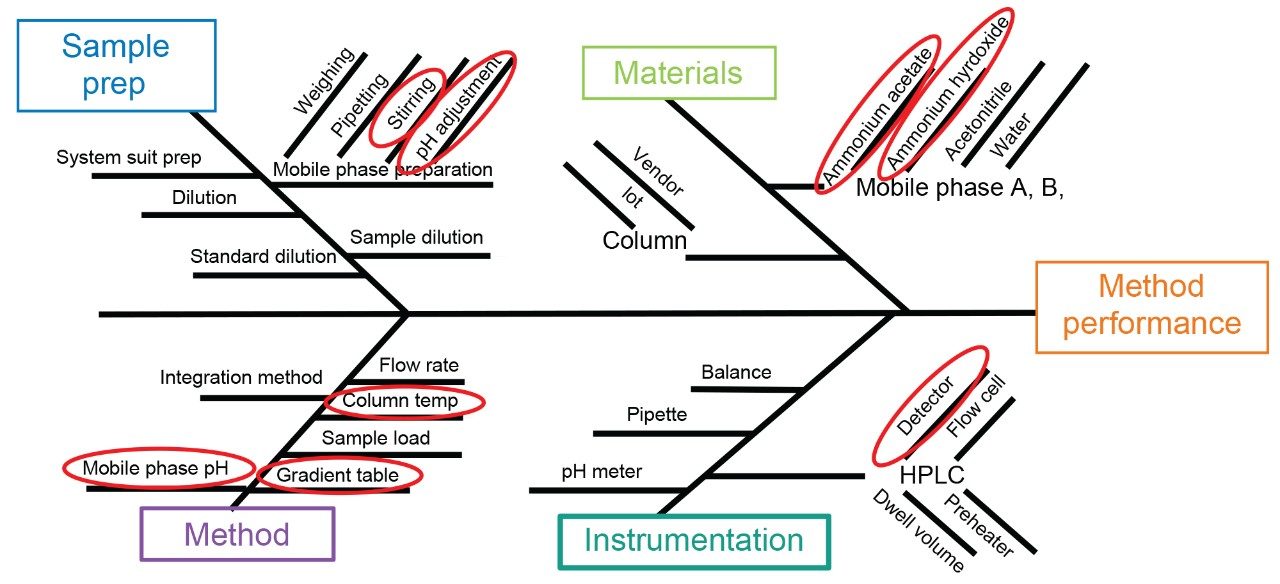
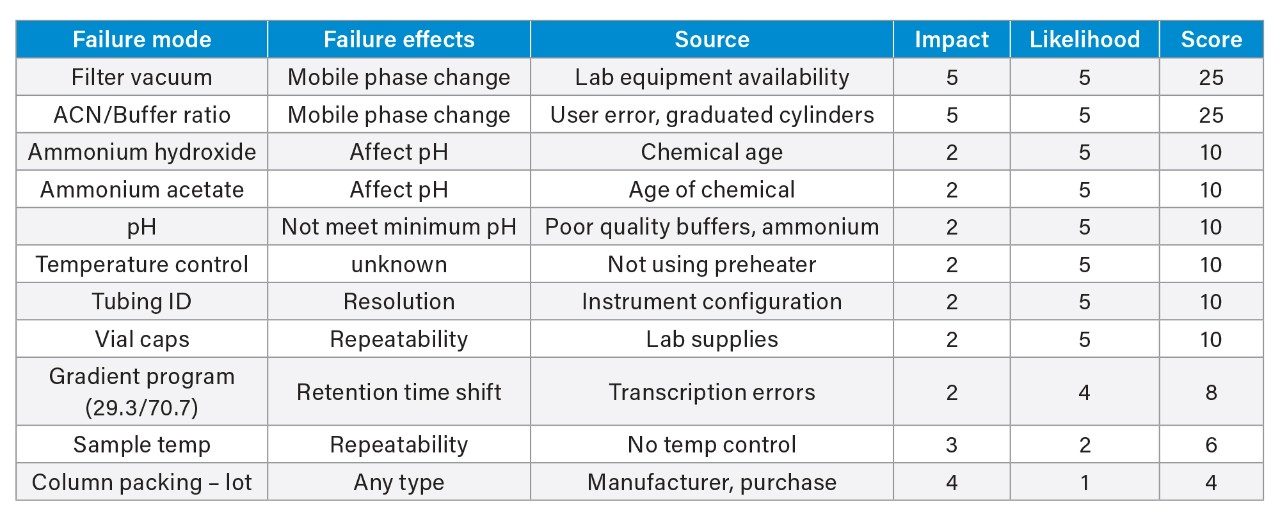
To reduce variability for the method transfer study, control strategies were developed based on the results from the risk assessment. These included providing the critical materials and SOP to the receiving lab from the sending lab. A single chemical kit was sent out to all the receiving laboratories and included the standards, the column, and the drug substance. The standards were purchased from the supplier (USP) in a single purchase and the drug substance was a single lot. However, the columns included two different lots. The SOP was written at the sending laboratory and then sent to a second site in Milford, MA for review, comments, and final approval. To ensure each lab was able to replicate the method - one which was unfamiliar to them - a detailed SOP was written. To ensure the instructions were comprehensive and clear, the second site in Milford acted as a beta site and reviewed and provided comments on the SOP. Within the SOP, specific instructions were also implemented to control lab to lab variability. For example, a preheater was required to reduce the impact of lab-to-lab temperature variability.
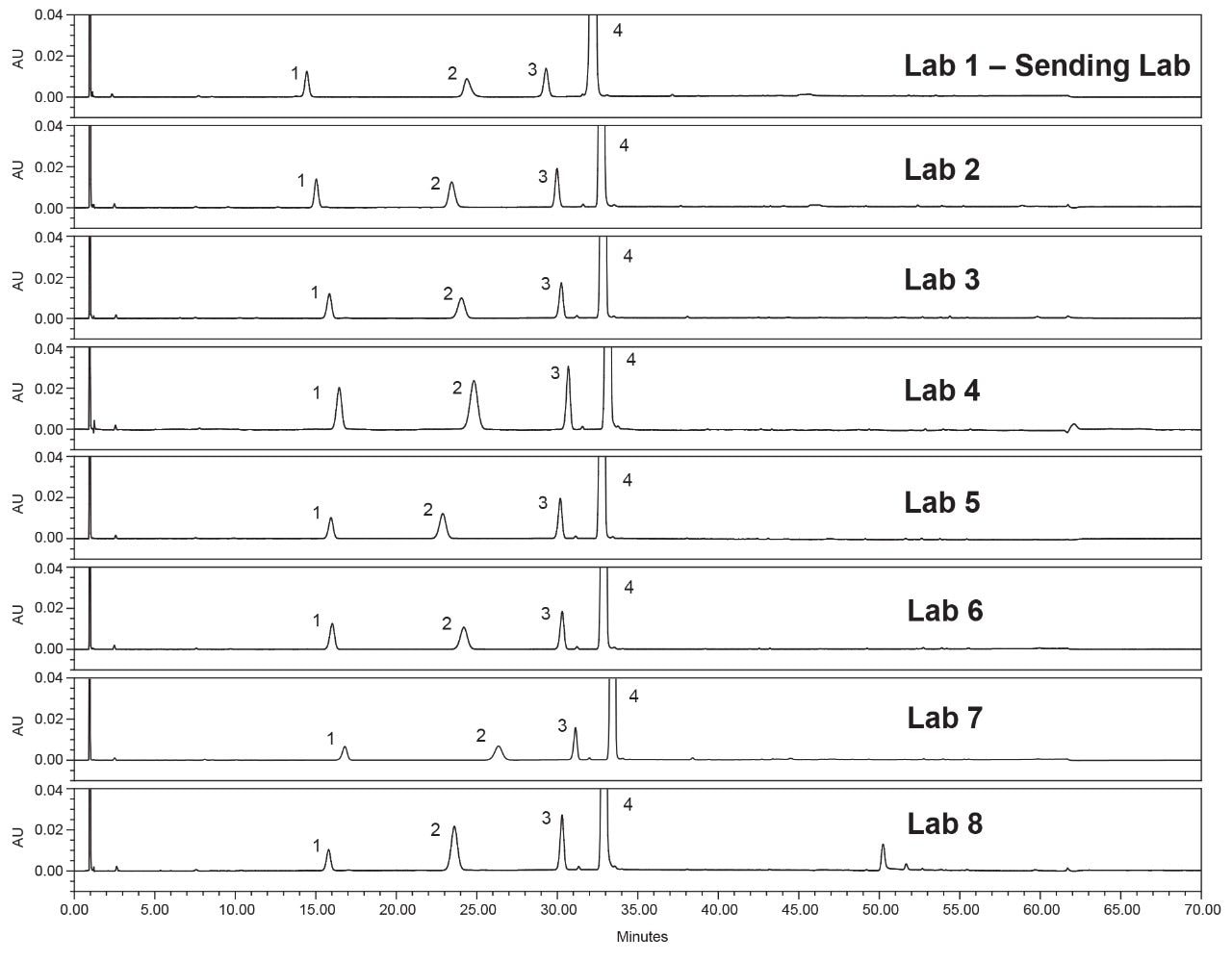
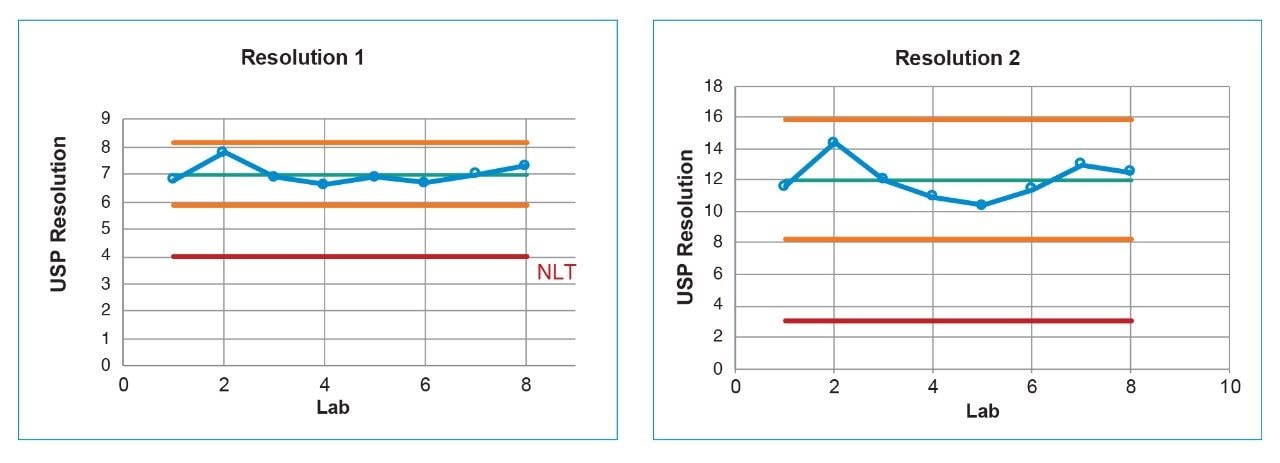
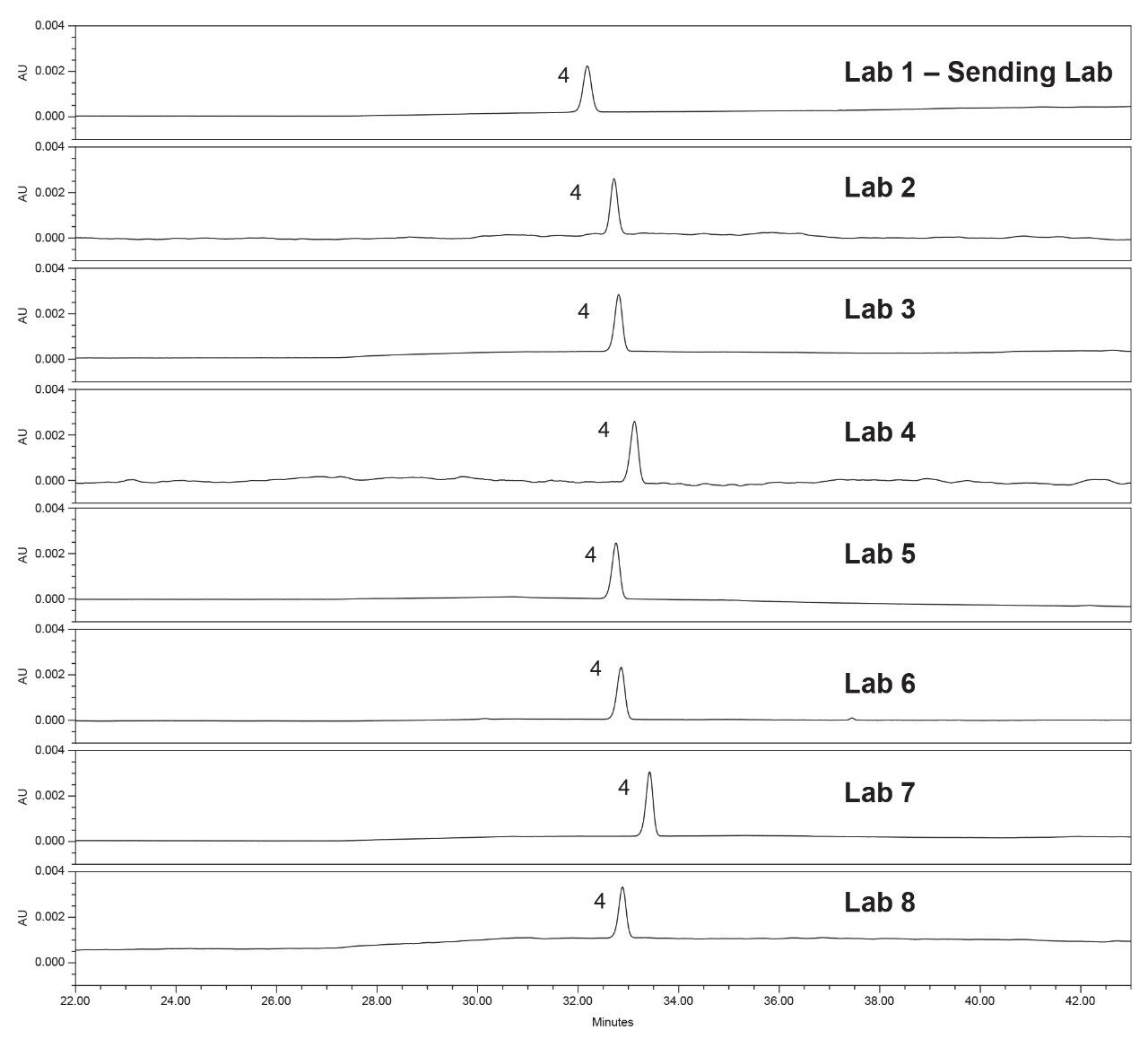
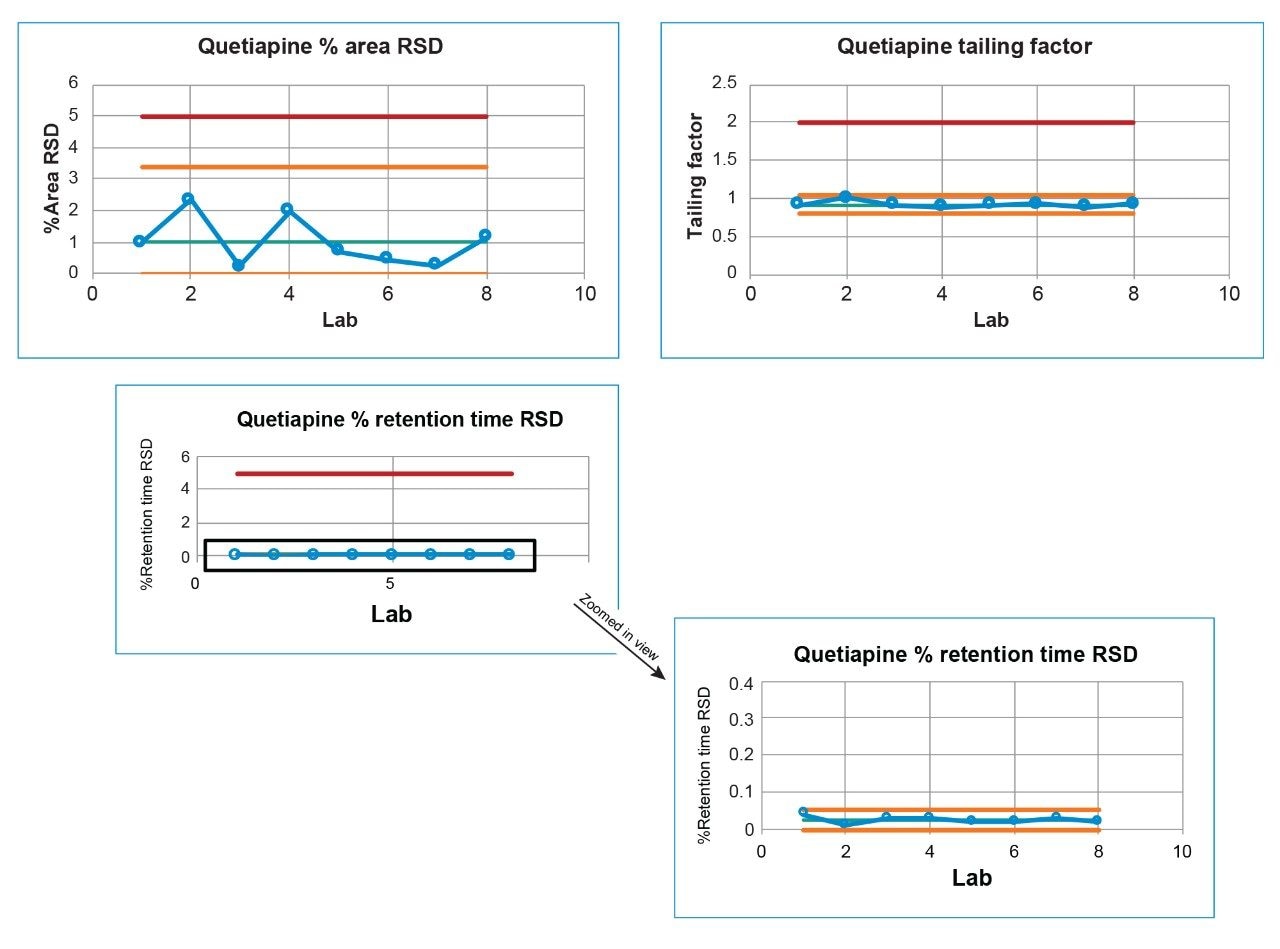
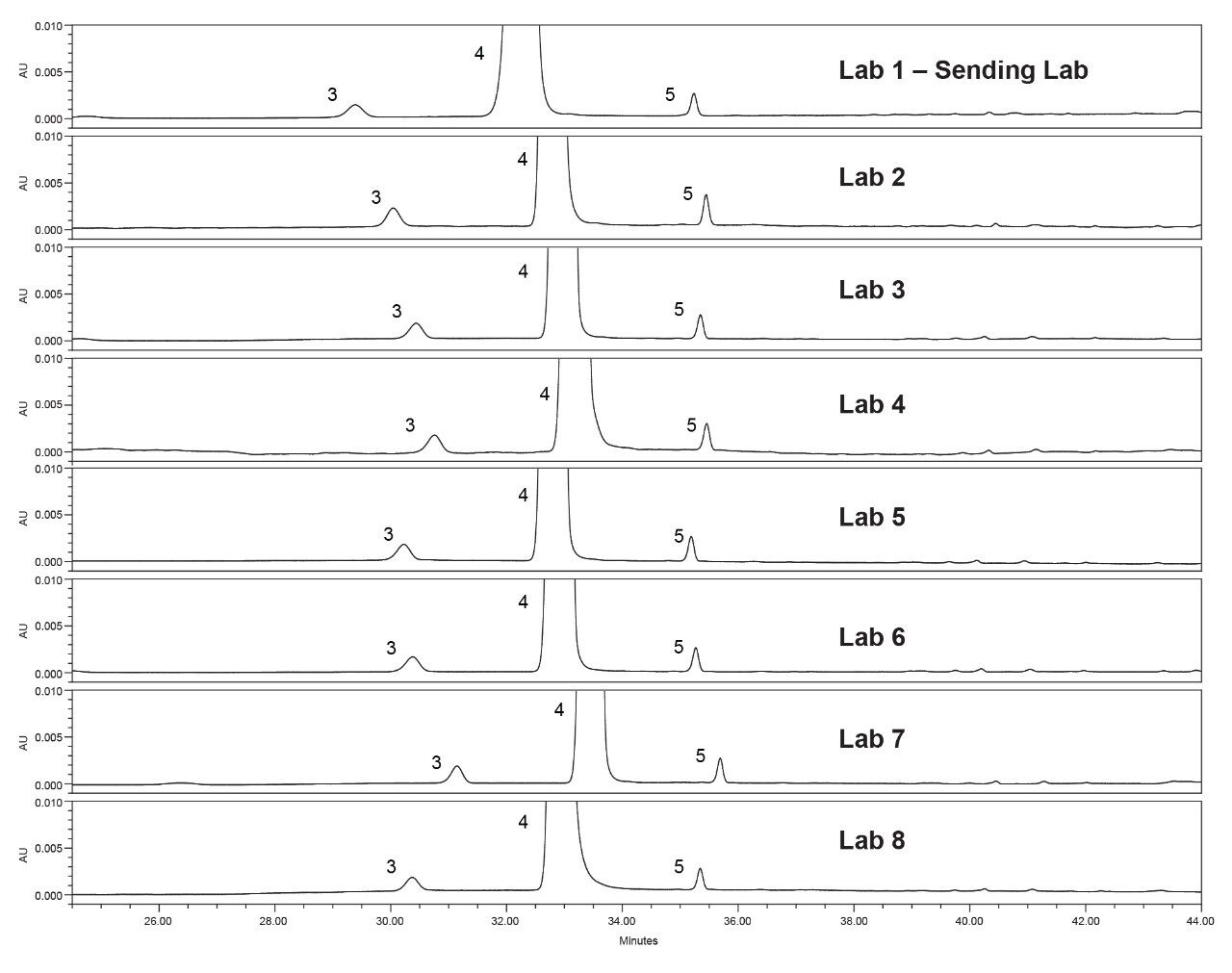
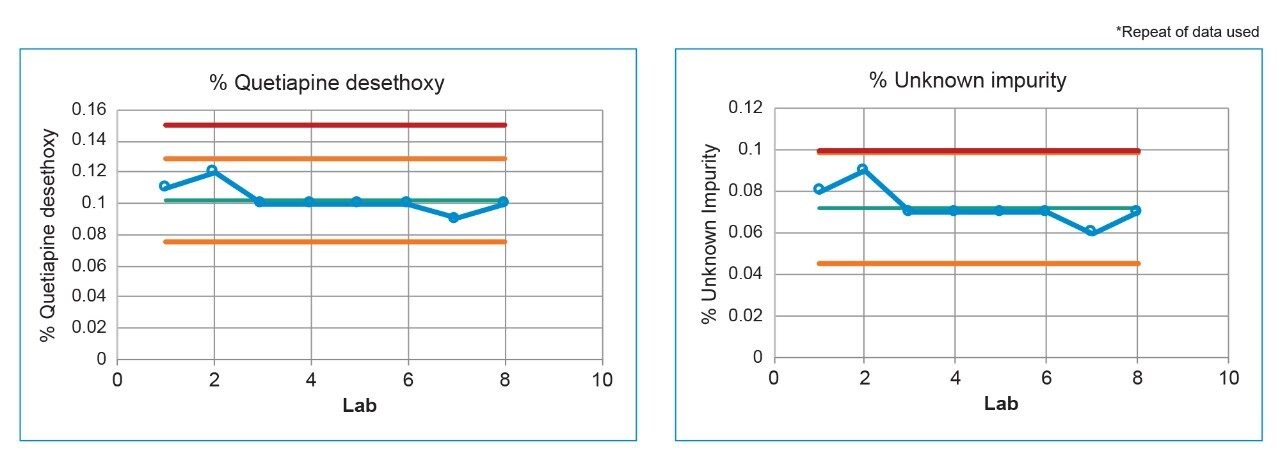
Lastly, each lab processed the system suitability analyses with a processing method provided within the SOP. However, all data presented in this publication was processed at the sending laboratory site to reduce variability in results due to different processing method parameters.
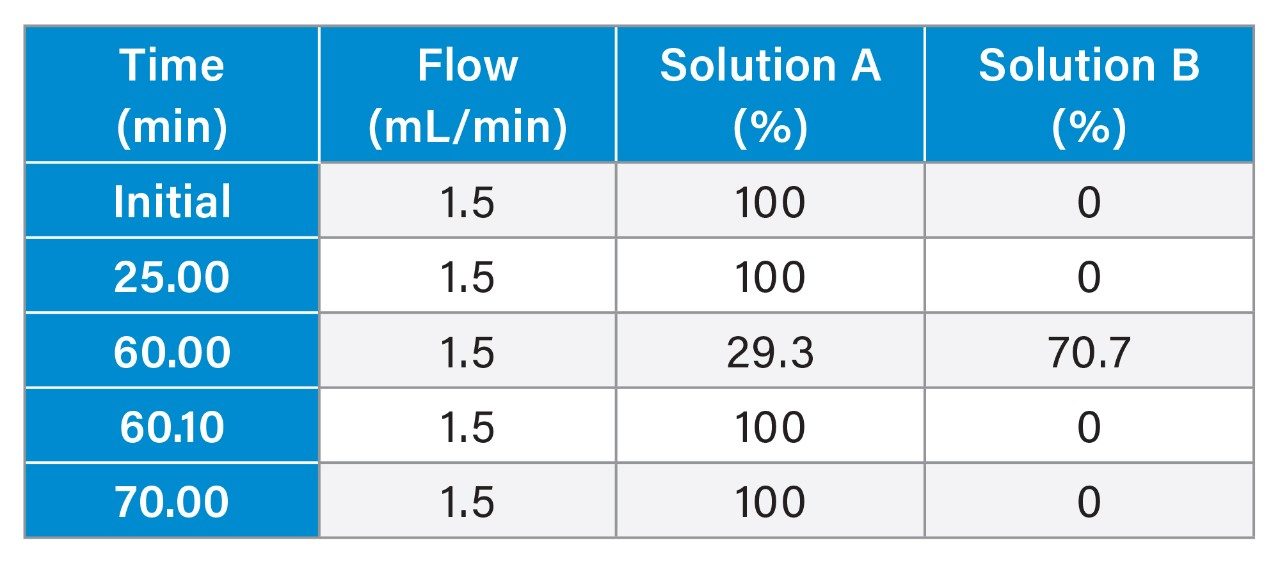
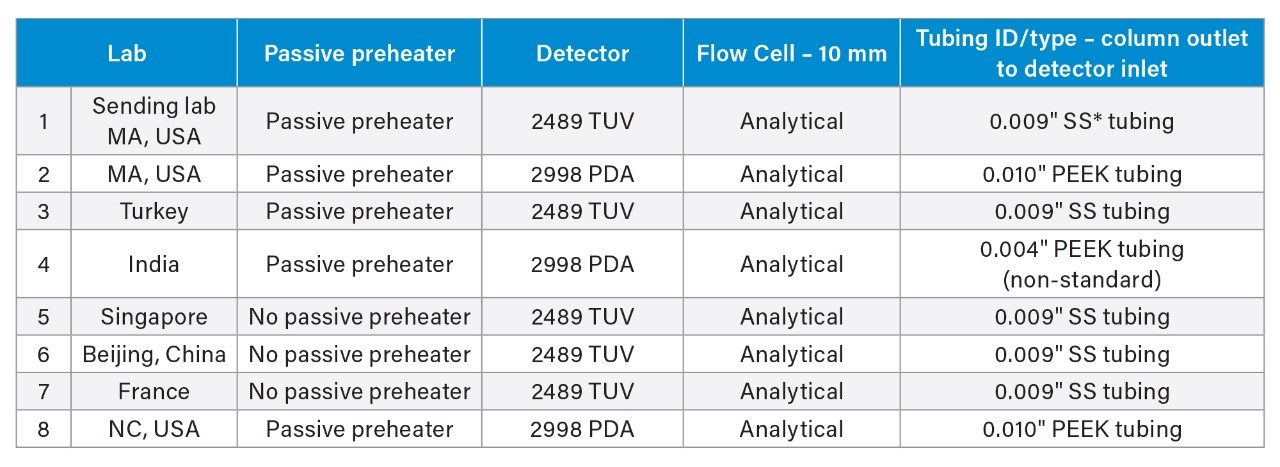
System configuration was also found to impact method performance. To control the system configuration at each site, specific configurations were requested in the provided SOP. However, due to availability of parts, some labs did not have the configuration requested. The receiving laboratories therefore recorded the configuration differences. The important aspects of the system included whether or not a passive preheater was used, the detector (TUV or PDA), the flow cell type, the flow cell path length, and the tubing ID from the outlet of the column to the inlet of the detector. The system configuration from each lab is listed in Table 2.