Increasing the Productivity of Oligonucleotide Purification through Column Scaling and Method Optimization
For research use only. Not for use in diagnostic procedures.
This is an Application Brief and does not contain a detailed Experimental section.
Abstract
For years, small molecules and protein-based therapeutics have dominated the pharmaceutical industry. However, there are limitations on the number of druggable targets and treatments that these modalities can address. Alternatively, synthetic oligonucleotides can be considered, especially when and where the transcription, splicing or translation of genetic information is implicated. Therapeutic oligonucleotides are a class of small synthetic nucleic acid polymers in the range of 20–30 mer that can be used for gene expression modulation. During drug development, from early discovery through to late stage development, there is a need for enough purified material to facilitate a range of different studies. Moreover, polymerase chain reaction and quantitative genomics approaches rely on oligonucleotides as molecular tools. This industry continues to expand with new innovation and is also in need of improved purification schemes. In this work, we present a cost-effective, high throughput, and systematic approach for both analytical separations and a preparative purification scheme for a 20-mer oligonucleotide.
Benefits
- Less hazardous, more affordable mobile phase additives that that can easily be removed after fractionation
- Scaling of an analytical UPLC separation to a widebore purification column
- Batch test and selected Oligonucleotide BEH™ C18 stationary phases to ensure reproducible separation and purification performance
- Optimization of chromatographic method parameters for high efficiency separations and purification
- Optimization of preparative on-column loading and fraction collection to maximize purity and recovery
Introduction
Efficient purification of synthetic oligonucleotides is often a challenging task. To date, it has been a challenge for chromatographers to select suitable columns, choose mobile phases, and optimize gradient conditions. Numerous publications describe the use of separations for the analysis and characterization of oligonucleotides.1–3 Analytical methods are frequently optimized through the use of high pH and elevated temperatures. The selection of a stationary phase must account for these chromatographic needs. Bridged-ethylene hybrid silica (BEH) C18 sorbents are selected for this purpose by many scientists. BEH C18 sorbents columns are available from Waters in analytical and preparative scale dimensions as XBridge C18 columns. Batch testing and selection of BEH C18 particles using oligonucleotide separations is additionally applied to produce ACQUITY™ and XBridge™ Oligonucleotide BEH C18.
Liquid Chromatography of oligonucleotides is predominantly performed by means of a Reversed-Phase Ion-Pairing (RP-IP) separation wherein alkylamine additives are used in the mobile phase and buffered with either acetic acid or with hexafluoroisopropanol (HFIP). Triethylamine (TEA)/HFIP aqueous mobile phases are the gold standard for oligonucleotide LC MS. Excellent peak shapes are obtained with this type of mobile phase, and the use of HFIP in place of acetate reduces ion suppression such that eluting oligonucleotides can be more readily detected by mass spectrometry.
One challenge of scaling oligonucleotide separations to a preparative scale is that HFIP is costly and hazardous. Laboratories can benefit from the reduced cost and mitigated exposure to HFIP by selecting alternative chromatography conditions for routine preparative scale separations. Secondly, small particle size sorbents are not usually applicable in preparative separations due to the high pressures they can generate. An attractive solution to this problem is to use reduced flow rates and columns packed with fully porous 2.5 µm particles. Columns for lab-scale preparative purifications in 4.6 mm, 10 mm, and up to 50 mm internal diameters (IDs) are commercially available from Waters. The optimal bed density (OBD™) packing technology utilized for preparative columns, enhances high throughput purification due to column lifetime related to the mechanical strength, reproducibility, and efficiency desired in preparative work.1
The use of a mobile phase based on triethylammonium acetate (TEAA) or hexylammonium acetate (HAA) can replace HFIP in the chromatography.2 Both TEAA and HAA are volatile and can be removed from the collected oligonucleotide fraction during evaporation or lyophilization. It has been shown that various alternative alkylamines with increased number of carbons can be used as ion-pairing reagents for separation of oligonucleotides.2 Selection of an alkylamine will be influenced by the properties of the oligo being purified. However, increasing the carbon length usually decreases volatility, increasing both the time taken and energy cost of the evaporation or lyophilization. Using methods with volatile mobile phases and additives is critically important for reducing costs, ensuring laboratory safety, and limiting the number of purification or post-purification steps.
In this application note, we demonstrate the scale-up of oligonucleotide purification methods, starting with an analytical UPLC™ separation based on TEA/HFIP mobile phase and ending with an HFIP free, widebore HPLC purification method.
Experimental
LC Analysis of 20 mer Oligonucleotide Model Mixture
Sample preparation
A crude 20-mer, 5’-G-C-C-T-C-A-G-T-C-T-G-C-T-T-C-C-A-C-C-T-3, was purchased from Oligo Factory, Holliston, MA for use as the probe oligonucleotide compound for this study. Sample solvent was prepared by reconstituting the crude mixture in 10 mM NH4OAc to a total concentration of 10 µM (59 µg/mL).
Mobile phase preparation
A mixture of 8.6/100 mM TEA/HFIP was prepared as mobile phase A. MeOH was used as mobile phase B. For other experiments, 100 mM TEAA pH 7.0 was prepared and used as mobile phase A along with acetonitrile as mobile phase B.
Columns
XBridge Oligonucleotide BEH C18 130 Å 2.5 µm Columns are employed in this studied. These columns contain batches of BEH C18 stationary phase that have been specially tested and lot selected for their oligonucleotide separation performance. QC testing performed in our Taunton, MA, USA ensures that only stationary phase batches capable of producing sharp, symmetrical peaks under ion pairing conditions are allocated for use in oligonucleotide columns. This is an important consideration for chromatographers to appreciate when looking to achieve and maintain the most reproducible methods and purification techniques.
Initial Analytical Method
|
Mobile phase A: |
8.6/100 mM TEA/HFIP |
|
Mobile phase B: |
MeOH |
|
Column: |
ACQUITY UPLC Oligonucleotide BEH C18 2.1 x 100 mm, 1.7 μm (p/n: 186003950) |
|
Flow rate: |
0.4 mL/min gradient |
|
Cool temperature: |
60 °C |
|
Diluent: |
10 mM NH4OAc in Water |
|
Sample con:. |
10 μM (59 μg/mL) |
|
Injection volume: |
2 µL (120 ng on column) |
|
Gradient: |
5% B for 0.25 min, 5–15% over 10 min, 15 95% over 0.25 min, hold 95% 1 min, 95–5% in 0.1 min, hold 5% 3.4 min. |
Optimized Analytical TEAA Method
|
Mobile phase A: |
100 mM TEAA pH 7.0 |
|
Mobile phase B: |
Acetonitrile |
|
Column: |
XBridge Oligonucleotide BEH C18 130 Å 2.5 µm 4.6 x 50 mm (p/n: 186003953) |
|
Flow rate: |
0.8 mL/min gradient |
|
Col temperature: |
25 °C |
|
Diluent: |
100 mM TEAA pH 7.0 |
|
Sample conc:. |
1 mg/mL |
|
Injection volume: |
10 µL (10 µg on column) |
|
Gradient: |
1–7.8% over 0.5, 7.8–9.8% over 10 min, 9.8–19% over 0.1 min, hold 19% 1.4 min, 19–1% in 0.1 min, hold 1% 4.9 min |
Optimized Preparative TEAA Method
|
Mobile phase A: |
100 mM TEAA pH 7.0 |
|
Mobile phase B: |
Acetonitrile |
|
Column: |
XBridge Oligonucleotide BEH C18 130 Å OBD Prep 30 x 50 mm 2.5 µm (p/n: 186008963) |
|
Flow rate: |
25 mL/min gradient 40 mL/min cleanout and equilibration |
|
Col temperature: |
25 °C |
|
Diluent: |
100 mM TEAA pH 7.0 |
|
Sample conc:. |
10 mg/mL |
|
Injection volume: |
42 µL, then varied |
|
Gradient: |
1–7.8% over 0.2, 7.8–9.8% over 12 min, 9.8–19% over 0.1 min, hold 19% 0.9 min, 19–1% in 0.1 min, hold 1% 2 min |
Results and Discussion
Switching Away from HFIP on an Analytical Scale
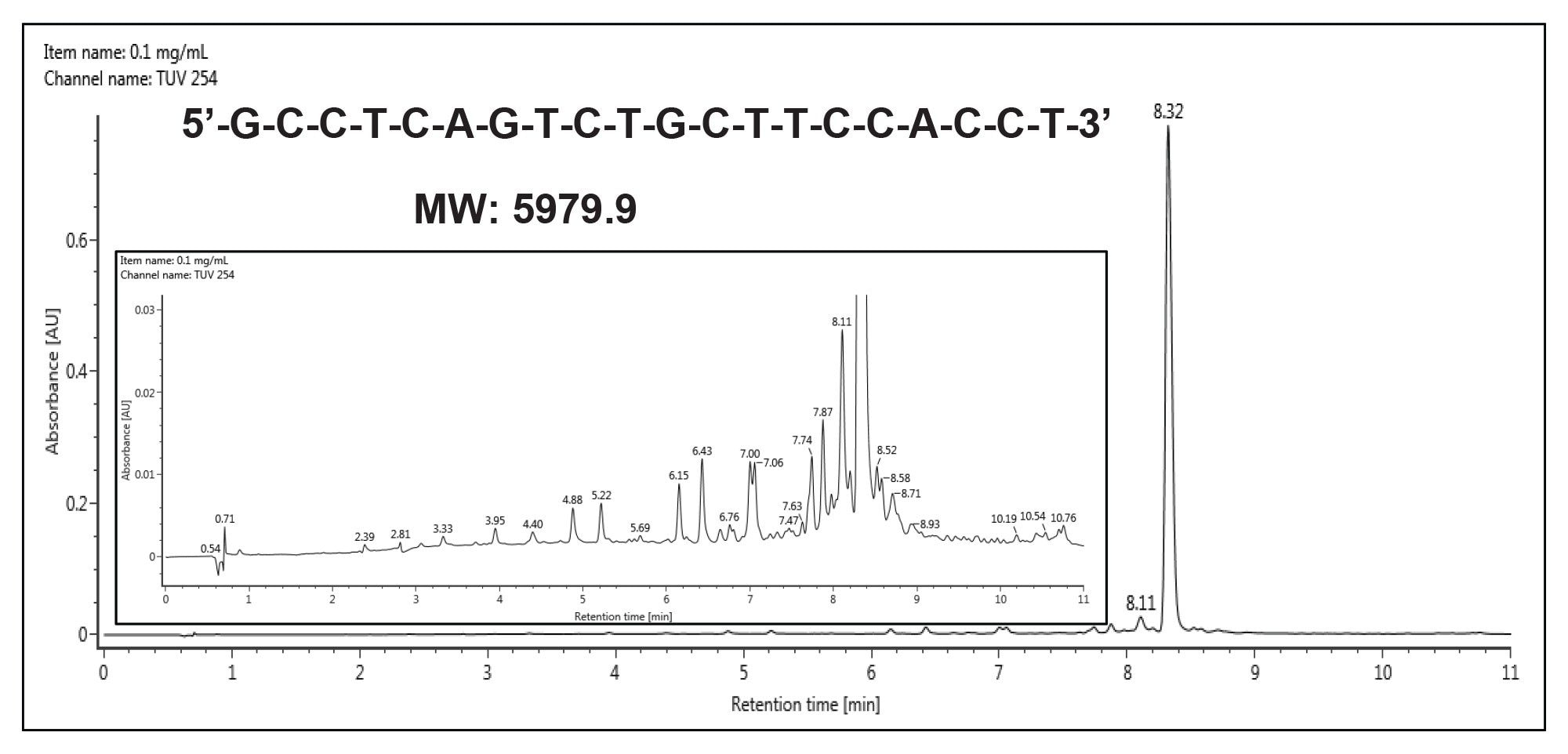
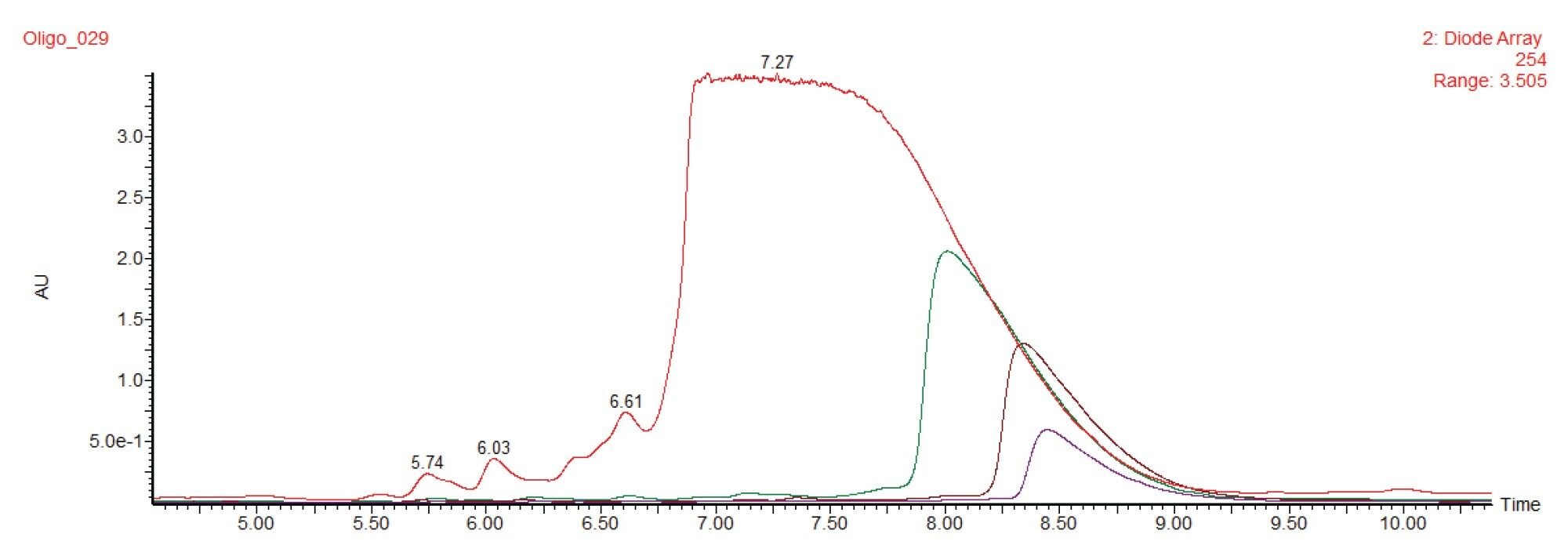
Separations of the 20-mer oligonucleotide were first performed on the analytical scale with an ACQUITY UPLC Oligonucleotide BEH C18 1.7 µm 2.1 x 100 mm Column. This method employed HFIP as part of the mobile phase and utilized a column temperature of 60 °C. Figure 1 provides an example chromatogram obtained with these conditions. A column temperature of 60 °C was used to eliminate an impact of secondary structure and to enhance the diffusivity of the oligonucleotide. As most short oligonucleotides lack secondary structures, they can be separated sufficiently well at ambient temperature which greatly simplifies the scale up to preparative scale.
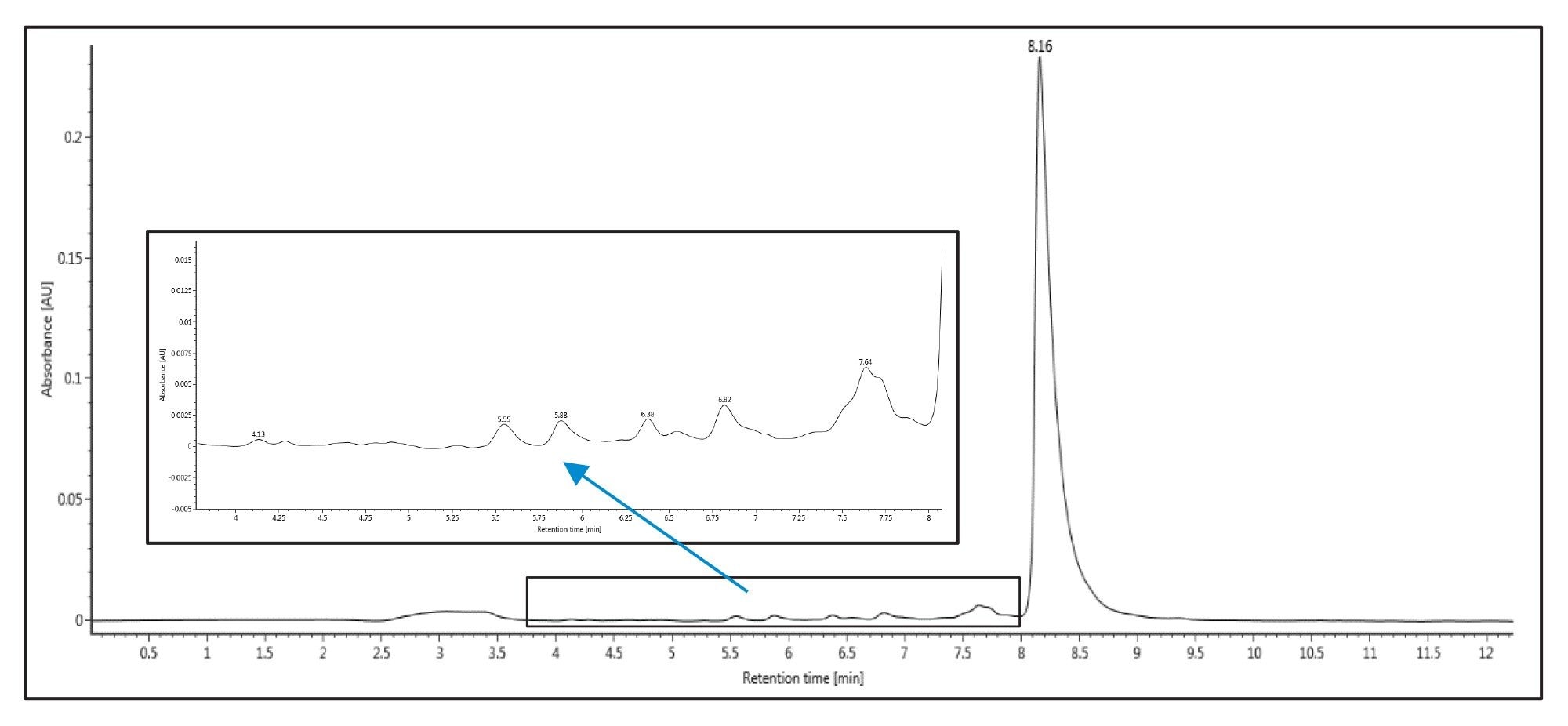
In the next round of experiments, a method using triethyl ammonium acetate (TEAA) was developed at the analytical scale. This method was performed using a 4.6 x 50 mm XBridge™ Oligonucleotide BEH C18 2.5 µm Column with mobile phase A comprising 100 mM TEAA pH 7.0 and mobile phase B made of acetonitrile. The column was maintained at 25 °C and the oligonucleotide was eluted with a multi-step gradient with a starting composition of 5% B. The composition was then increased from 5–8% B over 0.25 min, followed by 8–12% B over 9.75 min, before finally being increased from 12–95% B over 0.1 min. The composition was held at 95% B for 0.9 min, before returning 5% B in 0.1 min. The column was allowed to equilibrate at 5% B for 4.9 min prior to the next injection. Figure 2 presents the obtained chromatogram. Although the n-1 and n-x separation in TEAA is not as efficient as the initial TEA/HFIP run, this separation was still able to separate the target peak from the terminated failure sequences. This method provides a cost effective, less hazardous solvent system while maintaining solvent volatility for subsequent fraction collection and drying steps.
Scaling to a Preparative, Widebore Column
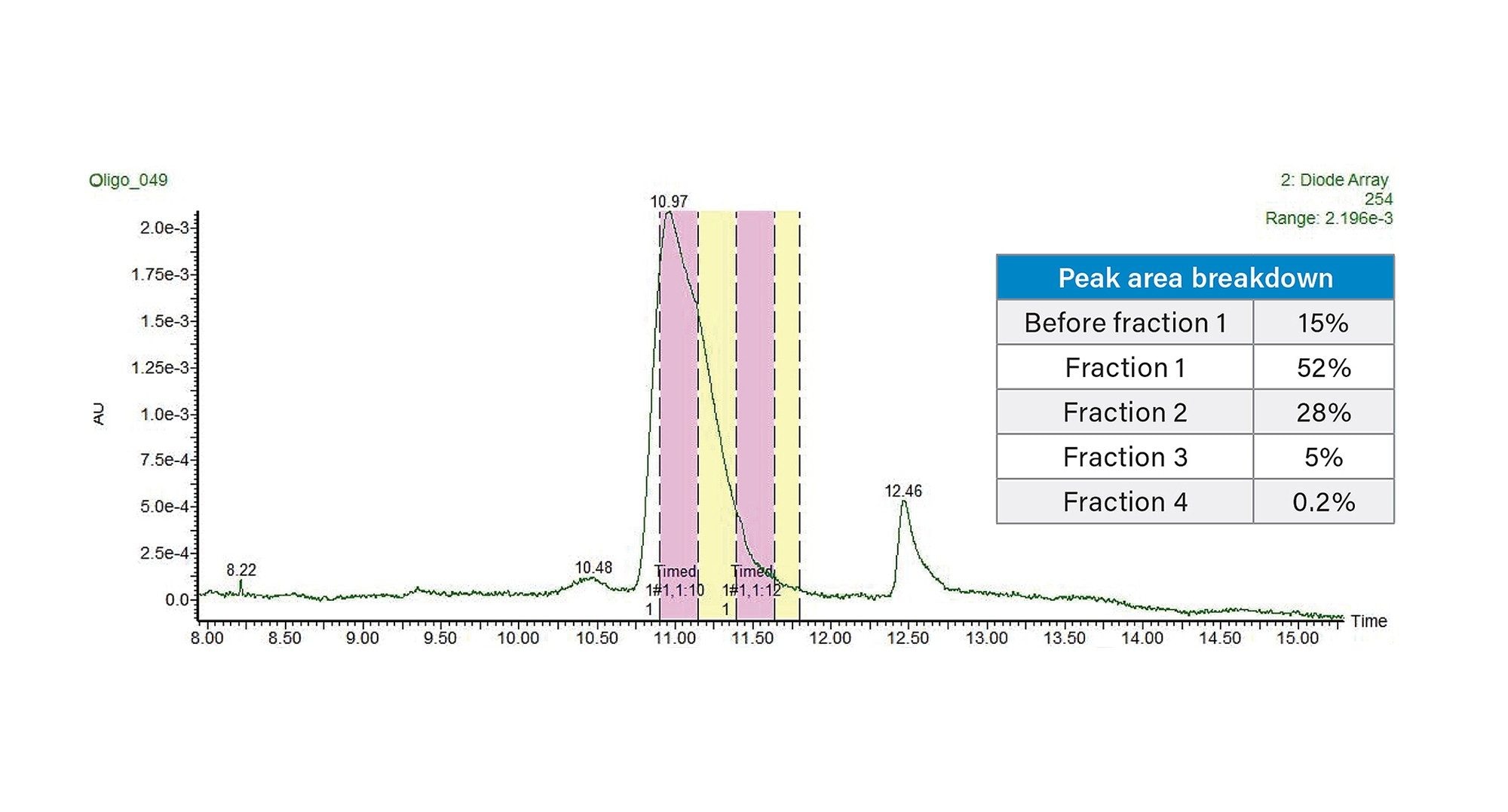
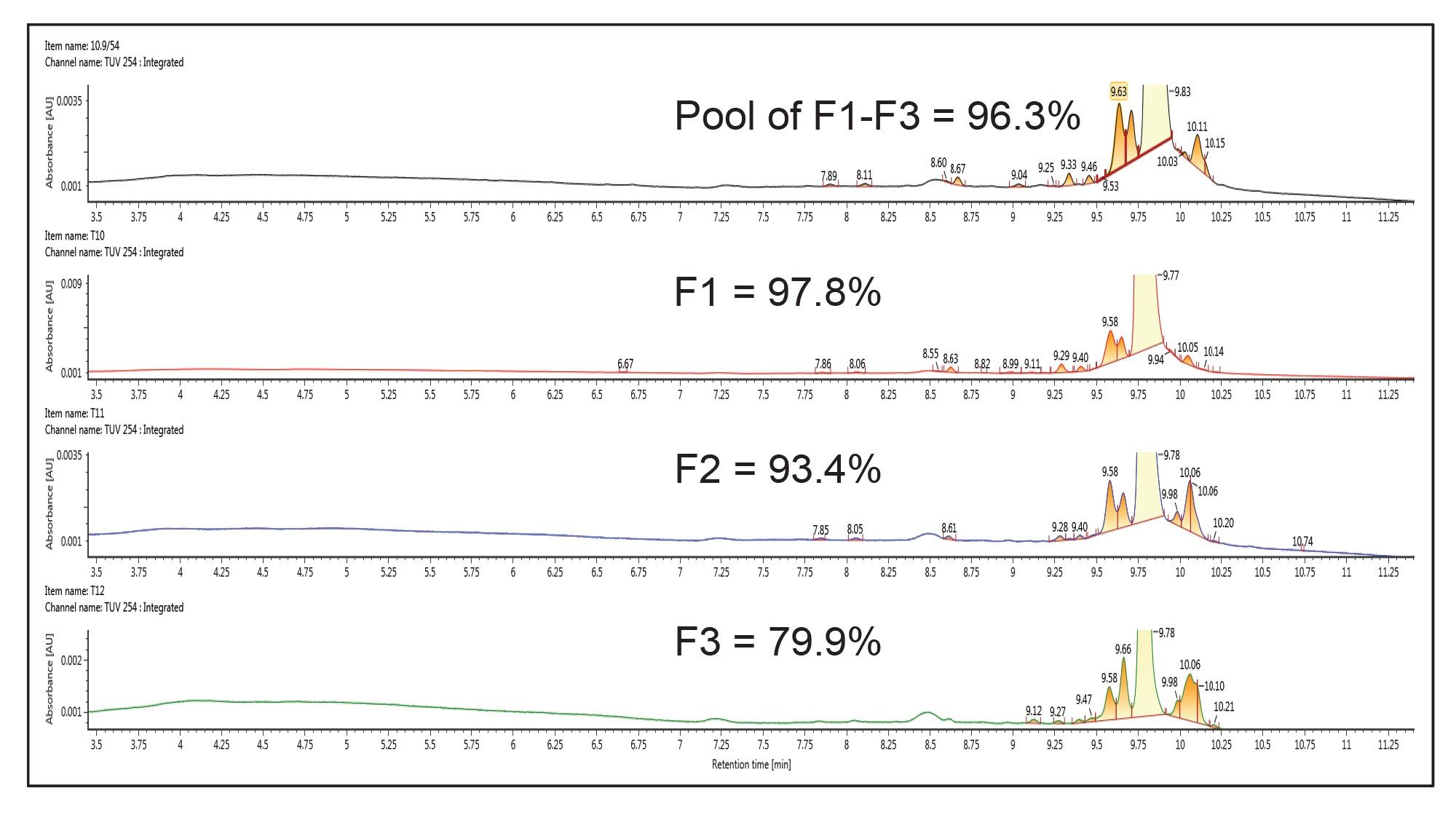
This new analytical method employing TEAA was then transferred to a 30 x 50 mm XBridge Oligonucleotide BEH C18 OBD Prep 2.5 µm Column using the Waters™ Gradient Calculator and Waters Autopurification System consisting of the 3767 autosampler/fraction collector, the SFO, a UV detector (PDA or TUV) and SQD2 single quadrupole MS and an ACQUITY ISM make-up flow pump for the MS. In this part of the work, the goal was to assess the purity across the peak, therefore a time-based collection was used. However, MS based fraction collection could be used and typically improves selectivity by collecting only the target mass. When scaling the method between different column IDs, one of the most important parameters to consider is the column flow rate that is proportional to the square of the column radius. In addition, for a scale up from the 4.6 mm inner diameter (ID) column to the 30 mm ID column, the scaled load factor is 42 times, based on the ratio of the column volumes. Therefore, the 10 µg analytical injection translates to a 410 µg preparative injection. A preparative scale chromatogram obtained at that mass load is shown in Figure 3. Fraction collection was triggered by using time, collecting 15 seconds of eluate per tube. Analysis of the collected individual fractions and the pooled fraction, using the initial UPLC method, shows that the individual fraction purity ranged from about 98% to 80% across the 3 major fractions (designated F1–F3 in Figure 3), with the pooled purity being 96.3%. The analytical purity analyses for these fractions are shown in Figure 4. The overall recovery of the pooled fractions was calculated to be 85%. Note, if increased purity is required, it would be recommended to not pool the third fraction. Pooled fraction purity for F1 + F2 would have been over 97% while recovery would have been impacted by about 5%.
Increasing Mass Load
The purification results shown in Figures 3 and 4 demonstrate that a robust analytical separation is scalable to a 30 mm preparative column without losing performance. However, another measure of a successful purification method is its productivity. The method produces ~1mg of purified oligonucleotide per hour. One option to improve productivity is to increase the mass load of each injection. The challenge with this approach is potential loss in resolution or recovery to obtain purified material, as a result of load dependent peak broadening. Figure 5 shows how chromatography on the 30 x 50 mm 2.5 µm oligonucleotide RPLC column is impacted as the mass load is increased up to 50 times X (25 mg). As expected, the peak liftoff moved further forward as the load increases.
Assessing Purity Across the Peak
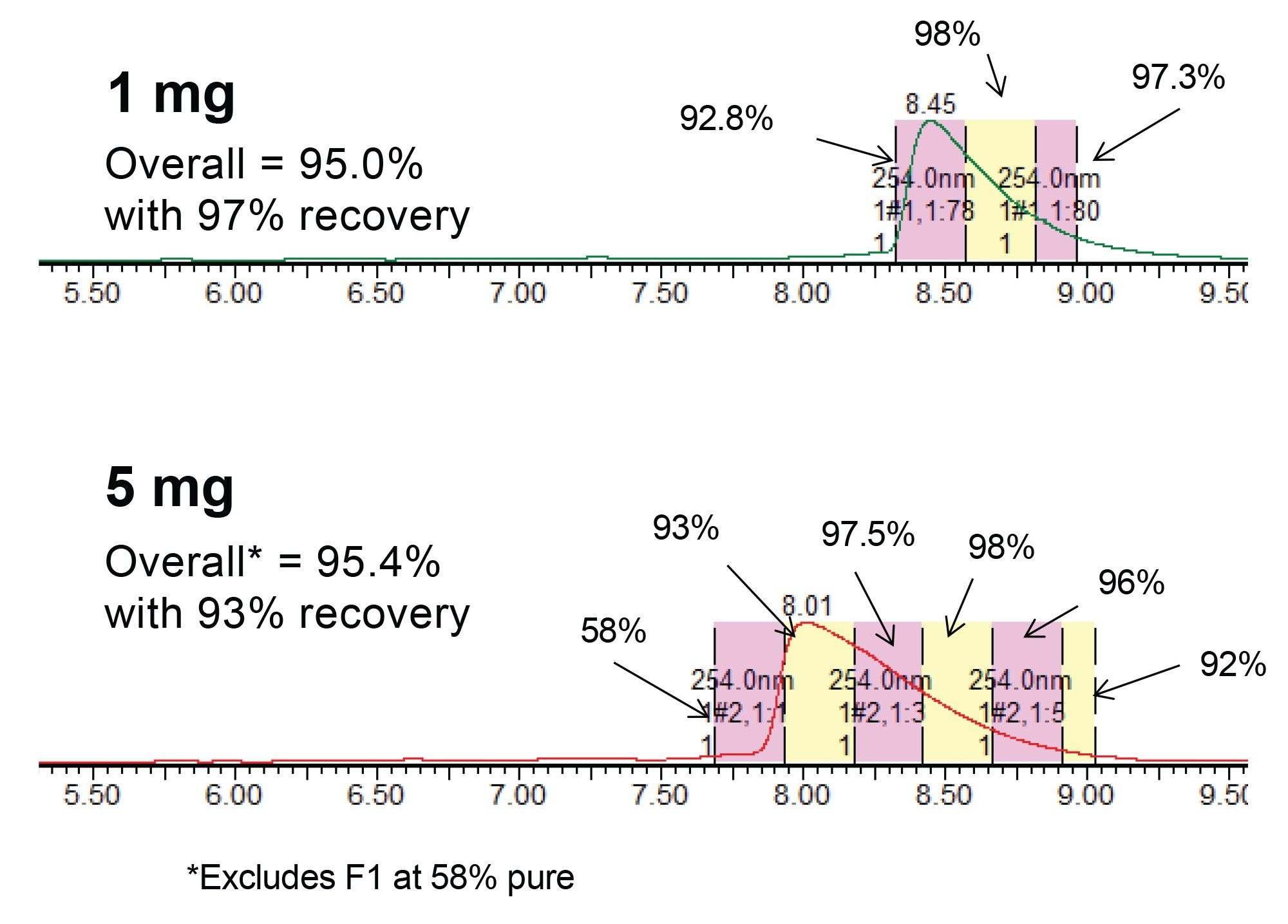
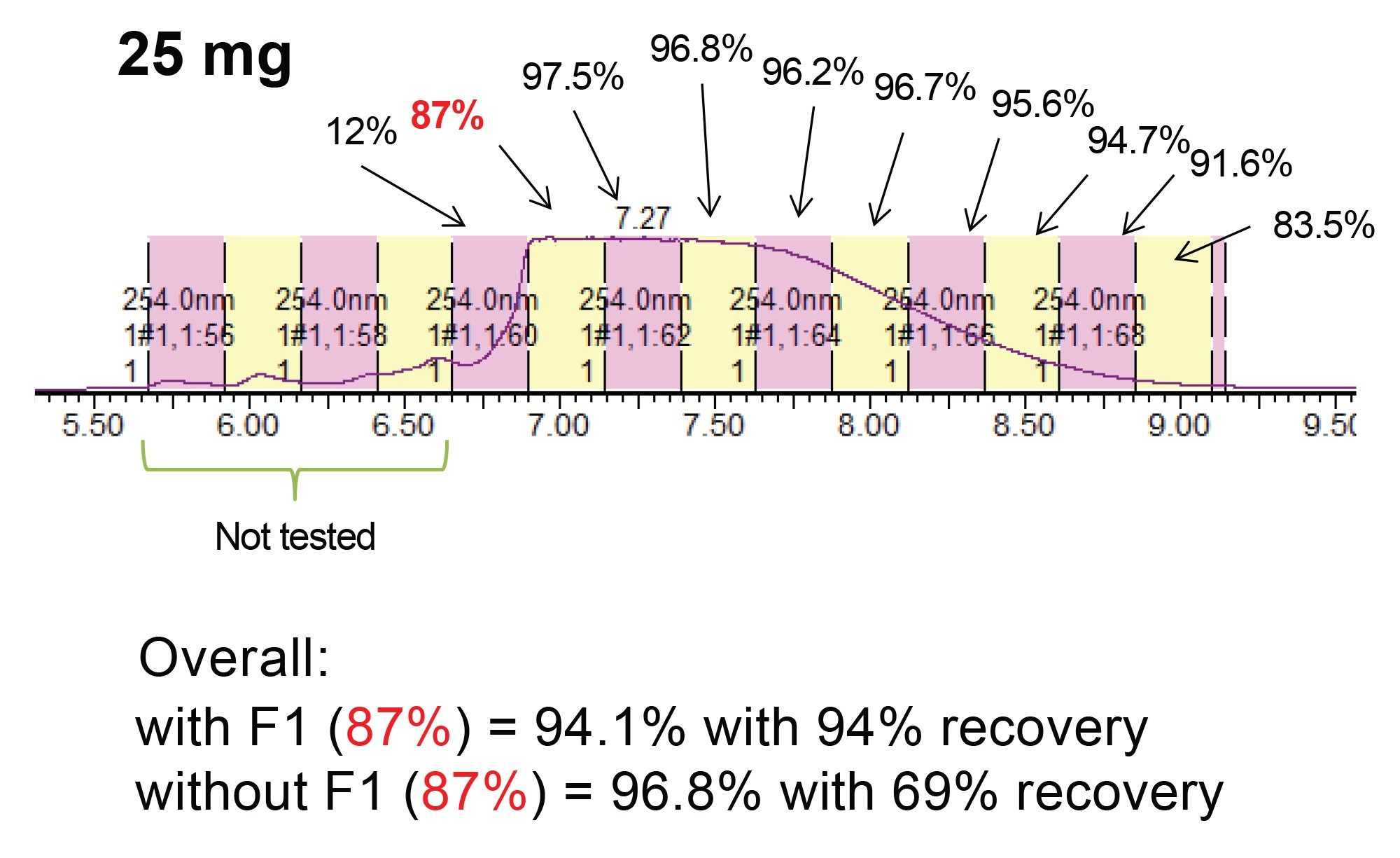
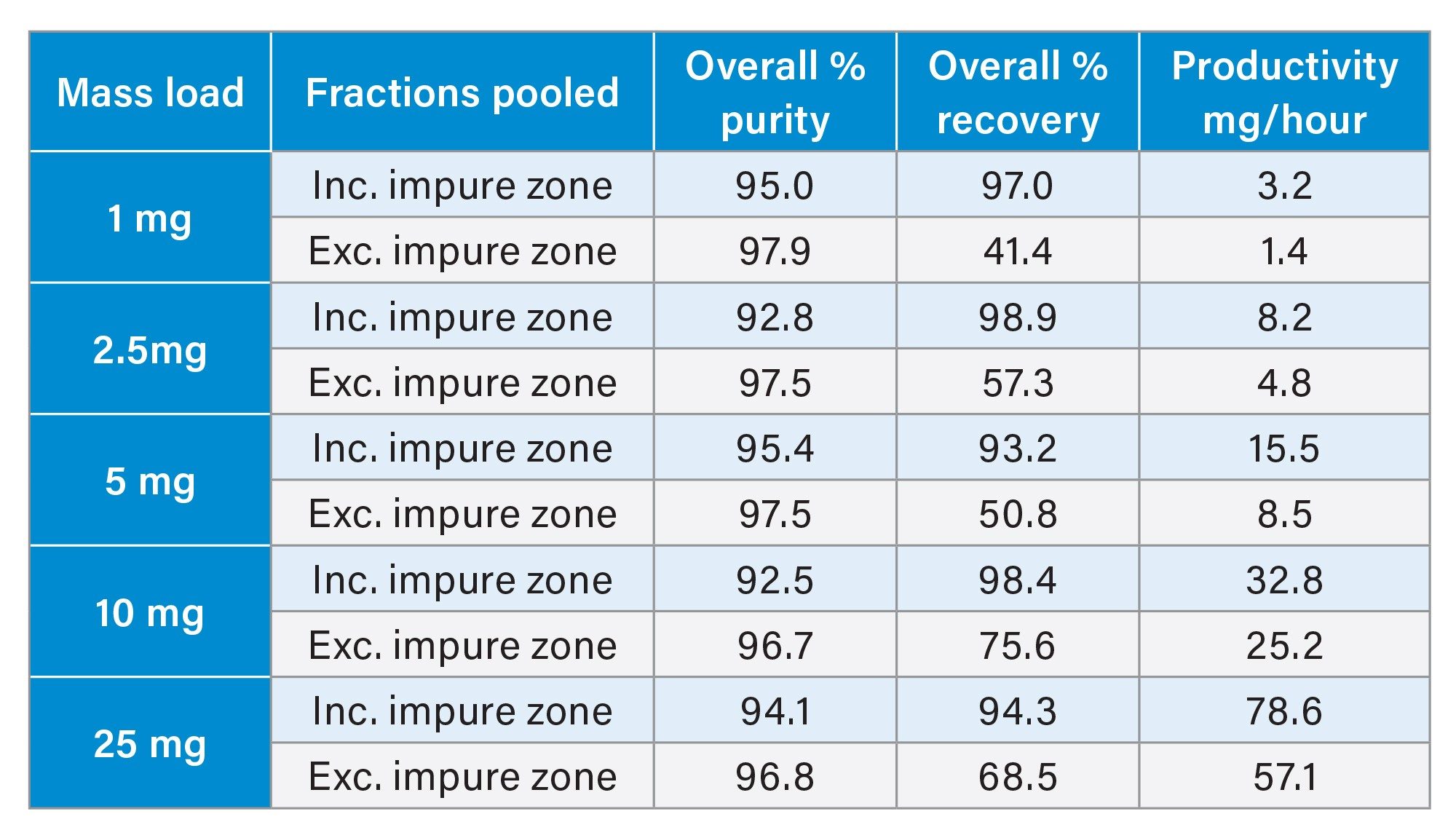
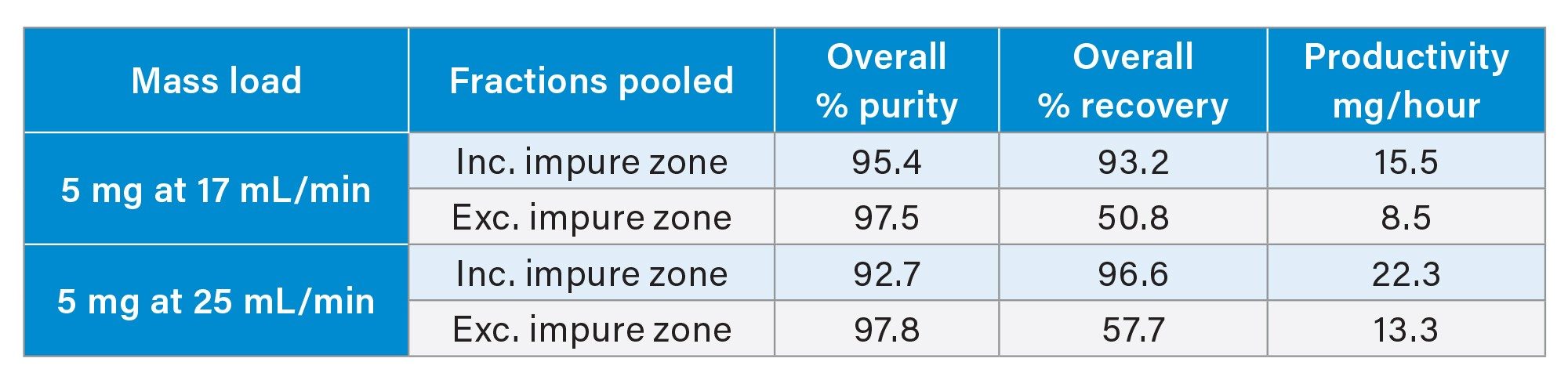
For each injection shown in Figure 5, fractions were collected in 15 second time intervals, concentrated and analyzed by the UPLC HFIP/TEA method. These purity determinations are provided in Figures 6 and 7. As previously reported the region of the peak consisting of the liftoff to the apex, is considerably less pure than other areas of the peak.3 However, as the peak broadens with increasing mass load, the number of pure fractions also increases, resulting in greater recovery and productivity. Based on the overall purity requirement, the necessary tubes can be pooled to generate the final purified material. Table 1 shows calculations on the productivity that can be achieved from the mass loads and varying fractionation approaches.
Method Parameters
Additional purification conditions were explored to further understand variables affecting productivity. Lower flow rates give improved separations but slower productivity. Increasing the gradient flow rate from 17 to 25 mL/min was assessed. Table 2 shows there is a slight drop in purity but a 50% increase in productivity.
Another approach to optimizing productivity is to apply a focused gradient. The previous examples already applied a focused gradient, so an increase to the flow rates outside of the elution window was investigated. Increasing the mobile phase flow rate during the regeneration and re-equilibration steps yielded a significant time savings.
Also, the method run time was slightly reduced to use the time while the autosampler is washing and injecting the next sample for the column to equilibrate. In this example, the run time was reduced by 1 minute. This combined with the flow rate modifications resulted in an overall increase in productivity of approximately 25%.4 The optimized preparative chromatography method is shown in Table 2. Based on the desired purity, this provides a method for generating purified oligonucleotides at ~75–100 mg/hour.
Finally, it is worth considering the use of other column dimensions, in particular an even wider bore column. This can provide an additional increase in productivity but was not tested here. Increasing from a 30 mm ID column to a 50 mm ID would provide an approximately three fold increase in load capacity and productivity, though it would require the use of a 47.2 mL/min flow rate. Yet another productivity enhancement could be made by implementing offline column cleaning and equilibration.4 This requires additional hardware including a switching valve, a regeneration pump and a second column. However, it completely eliminates this time from the method.
Conclusion
This application note shows the interplay and scalability between high efficiency analytical purity measurements and high productivity purification methods. While HFIP mobile phases are desirable for analytical purity LC-MS measurements, they become problematic for use with purification processes. As such, we have demonstrated the successful transfer of an HFIP mobile phase analytical separation to TEAA based purification runs on a widebore optimal bed density preparative column. Through the optimization of method parameters and mass load, a simple lab scale purification scheme was devised that produces over 95% pure material at a rate of over 100 mg/hour. Further increases could be realized by increasing the ID of the preparative column and implementing offline column cleaning and regeneration. It is our hope that these techniques will be of value to the industry as they take on new initiatives in support of the ever-expanding synthetic oligonucleotide industry.
References
- Patrik D. McDonald et al., Optimum Bed Density [OBD™] columns: Enabling Technology for Laboratory-Scale Isolation and Purification 720001939.
- Gilar, M. et al., Best Practices for Oligonucleotide Analysis Using Ion-Pair Reversed - Phase Liquid Chromatography -Columns and Chemistries 720006948.
- Gilar, M. et al., HPLC and UPLC column for Analysis of Oligonucleotides, Waters 720002376.
- Lefebvre, P. et al., Evaluating the Tools for Improving Purification Productivity. Waters application note 720001696. 2007
720008266, February 2024