方法开发
色谱柱筛选
要使用色谱方法分离目标化合物,必须先进行方法开发。通常先开发分析级色谱方法,目的是节省溶剂、时间和样品。对于SFC,我们无法预测混合物中各组分在指定固定相中的保留情况。不同于RPLC,选择合适的色谱柱填料对于SFC分离而言至关重要,而且这也是SFC方法开发中十分重要的因素。因此,我们要借助有效的筛选策略大幅提高纯化目标的分离效果7。目前,各生产商都在积极开发新型固定相,未来有望出现几种SFC分离理想的色谱柱填料。如今,大部分用户都会对适用于应用的“理想”色谱柱执行自动筛选7。
在选择适合的色谱柱用于筛选时,需要考量多个因素。由于分析型色谱柱的L/dp(柱长/粒径)需要与最终的制备型色谱柱保持一致,因此只有规格满足要求的分析型色谱柱才能纳入考虑范围。这样可确保色谱方法在系统间放大时的一致性。所选色谱柱应覆盖宽泛的选择性并适应样品特性。例如,强极性化合物(例如糖类)不太可能在C18色谱柱上保留,而硅胶色谱柱可能不是疏水性极强(非极性)的化合物(例如类胡萝卜素)的理想选择。还可以根据色谱柱对碱性化合物、酸性化合物或这两者的适用性来分类。
手性纯化需要使用手性色谱柱。由于手性色谱柱固定相与目标化合物之间的相互作用更加复杂,因此需要通过更多的试错实验来确定手性分离的理想色谱柱。不过,某些色谱柱的命中率高于其它色谱柱。例如,衍生自纤维素和直链淀粉的手性固定相以其广泛的适用性而闻名7,还有一些手性固定相(例如Pirkle型手性固定相)则以化学稳定性高而著称。手性固定相还适用于非手性应用,尤其是涉及位置异构体或密切相关化合物的应用3。
色谱柱筛选通常在梯度溶剂条件下进行,助溶剂比例范围一般为2%~50%。上述条件有助于用户准确判断哪种色谱柱适合分离目标化合物,还能很好地指示洗脱化合物所需的助溶剂比例。图17所示为(R)和(S)-告依春(一种发现于板蓝根根部的活性天然产物)分离的手性色谱柱筛选示例,共使用了五种纤维素和直链淀粉基固定相(IC、OJ-H、AS-H、OD-H和AD-H)和一种Pirkle型固定相(S,S)-Whelk-O116。图18为非手性色谱柱筛选示例,其使用三种类胡萝卜素组成的混合物对四种非手性固定相进行了筛选17。由图可知,类胡萝卜素在C18上保留效果好,在极性较强的其它固定相中保留效果较差。
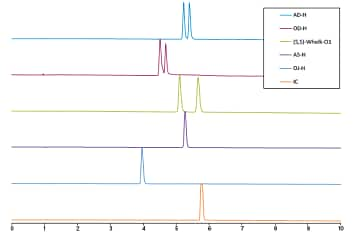
图17. 手性色谱柱筛选示例。六种不同固定相分离(R,S)-告依春标准品得到的SFC色谱图。所用梯度为:10 min内从5%升至40%,40%保持2 min,然后在1 min内从40%降至5%,5%保持2 min16
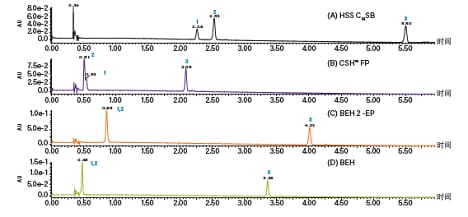
图18. 非手性色谱柱筛选示例。不同色谱柱分离番茄红素、ß-胡萝卜素和叶黄素混合物得到的UPC2UV色谱图:(A) HSS C18 SB;(B) CSH氟苯基柱;(C) BEH 2-EP;(D) BEH。峰标识为:1. 番茄红素;2. ß-胡萝卜素;3. 叶黄素。所用梯度为:5 min内从5%升至20%,20%保持2 min,然后在1 min内从20%降至5%17。
溶剂筛选
一般来说,仅使用CO2不足以将化合物从色谱柱上洗脱;因此通常需要添加极性助溶剂。助溶剂的选择是SFC色谱方法开发和优化的一个关键参数。大部分应用都可以使用多种助溶剂和混合溶剂来优化分离。虽然SFC兼容的溶剂范围看起来非常广,但我们可采用配对极性谱两端的溶剂这种方法来简化溶剂筛选工作;将非极性CO2与强极性有机溶剂配对,可得到各种溶剂强度的流动相。
四种最常用的助溶剂为甲醇、乙醇、异丙醇和乙腈;其中甲醇极性最强,乙腈极性最弱。一般建议在色谱柱筛选和溶剂初筛时使用甲醇或乙醇。异丙醇和乙腈适用于方法优化,或者使用甲醇/乙醇无法达到可接受分离度的情况。在很多情况下,组合使用这些溶剂能够起到微调流动相极性的作用。例如,向甲醇中加入乙腈可降低助溶剂强度,从而延长保留时间并改变选择性。保留时间通常会随助溶剂强度下降而延长(图19)。另外,与LC相同,保留时间还会随强溶剂比例的增大而缩短(图20)。不过,考虑到CO2粘度较低,有时也可通过提高总流速(助溶剂比例保持不变)来缩短运行时间,而不影响分离度(图21)。
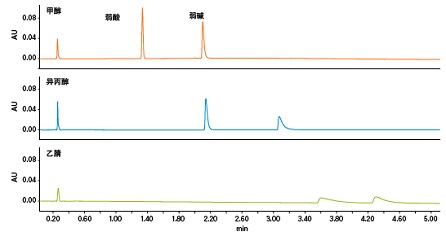
图19. 展示助溶剂强度影响的色谱图。
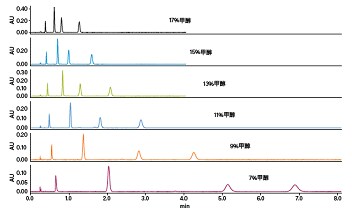
图20. 助溶剂比例对保留时间的影响。
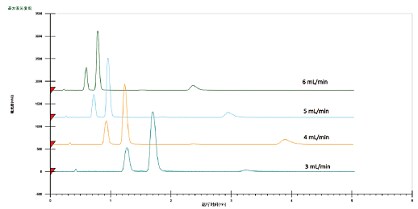
图21. 总流速对色谱分离的影响。
助溶剂中的添加剂
对于制备型色谱而言,获得尖锐的峰形和高分离度非常重要,峰变宽和流动相溶解性不佳会带来灾难性后果。向流动相的助溶剂部分加入添加剂可减轻SFC的峰拖尾现象,该方法可扩展基于CO2流动相适用的溶质范围10。图20. 助溶剂比例对保留时间的影响。
CO2作为色谱溶剂配合其它极性有机溶剂使用时,可得到略偏酸性的流动相。而正是由于基于CO2的流动相略偏酸性,在分析许多酸性和中性化合物时,无需使用流动相添加剂即可得到可接受的峰形。不过,酸性添加剂可进一步改善某些酸性化合物的峰形(图22)。常用的酸性添加剂包括三氟乙酸、甲酸和乙酸。涉及碱性化合物的应用一般需要使用碱性添加剂(添加量一般为0.1%~1%),其目的是改善色谱分析性能(图23)。常用的碱性添加剂为仲胺或叔胺,例如异丙胺(IPA)、二乙基胺(DEA)或三乙胺(TEA)。这些添加剂即使浓度极低(可低至助溶剂的0.01%),效果也非常明显,但它们会干扰MS检测,还可能导致某些色谱柱填料(尤其是未键合硅胶填料)出现记忆效应。在这种情况下,可考虑换用醋酸铵和氨水,它们对色谱柱和MS检测的兼容性更好,甚至还有助于强化MS信号。
除了传统助溶剂之外,许多新型色谱柱还兼容包括乙酸乙酯、四氢呋喃、二氯甲烷、氯仿或二甲氧基甲烷在内的其它非传统助溶剂或添加剂。组合使用这些溶剂还可进一步优化保留时间和选择性,同时改善样品溶解性⁷。水就是一种应用越来越广泛的非传统添加剂。水不与CO2混溶,但作为添加剂加入强极性助溶剂(例如甲醇)时却能有效提高流动相溶解性并改善亲水化合物的SFC峰形(图24),其允许添加量取决于助溶剂占总流动相的比例。最后,流动相发生的相分离(不混溶)会导致基线噪音10。一般极性助溶剂中水的有效添加量为1%-5%。使用水作为添加剂,Prep SFC可纯化极性更强的化合物³。
纯化时最好避免使用需要在下游去除的添加剂。如果分离时必须使用添加剂,应使用易于去除的挥发性添加剂,并按最低有效浓度添加。另外,现在已经出现了对添加剂依赖程度较低的新型固定相,这对Prep SFC非常有利。
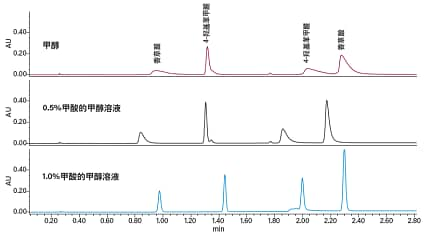
图22. 酸性添加剂对酸性化合物峰形和分离度的影响。
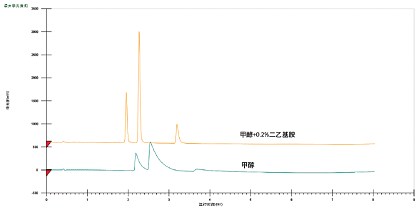
图23. 碱性添加剂对碱性化合物峰形和分离度的影响。
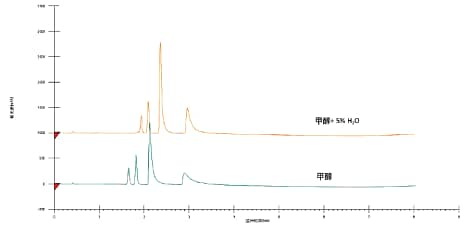
图24. 水作为添加剂对极性化合物分离的影响。
密度:温度和压力
所有化合物的溶解性和保留因子都与流体密度密切相关。流经色谱柱的流动相密度至关重要,因为这决定了流动相的理化特性⁵。SFC非常有趣的一个特性就是能够通过调节温度和压力来控制流动相密度。尽管色谱柱和流动相的选择对SFC分离的影响最大,利用温度和压力也可对分离进行微调或优化。在这两个参数中,压力对色谱分离的影响更大;随着压力增大,流动相密度也会变大,这通常会导致保留时间缩短和分离度下降(图25)。另一方面,升高温度则会使流动相密度变小,保留时间延长。不过,温度调节的效果受化合物限制,还会改变化合物分离度,甚至是邻近洗脱化合物的洗脱顺序;这种现象在手性分离中更常见(图26)。
压力控制由背压调节器(BPR)完成,该装置可设置色谱柱之后的系统“背压”。尽管系统被设计为可适应较广的压力范围(最高可耐受400 bar的设定值),但在Prep SFC应用中,一般将背压调节器设定为100~200 bar (1450~2901 psi),并利用柱温箱(加热色谱柱)或热交换器(加热流动相)将温度控制在设定值。温度设定值范围一般为40~60 °C。部分应用在亚临界(液态CO2)条件下运行,这种情况下的设定温度较低,一般为25~35 °C,同时需保持相对较高的压力。不建议在压力和温度接近临界值的条件下运行系统,因为此时即使温度或压力发生微小变化也会显著改变CO2密度(以及色谱分离结果)⁵。温度和压力的限值通常与色谱柱填料或装填方式的稳定性有关。
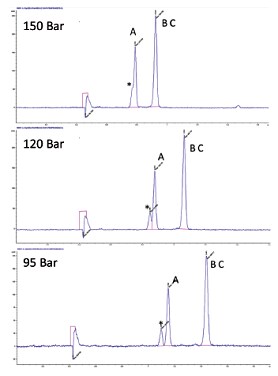
图25. 压力对保留时间和分离度的影响。
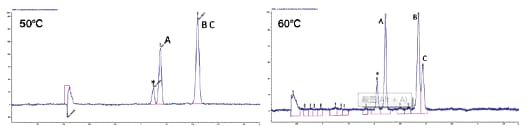
图26. 温度对保留时间和分离度的影响。
优化流动相条件
理想的溶剂和色谱柱组合通常根据应用或目的而异。理想情况下,大上样量、高分离度以及快速的分离相结合可完全回收高纯度的目标化合物。但在实际应用中通常需要权衡,找出能够实现生产效率高效优化的解决方案。例如,虽然(采用大上样量时)色谱峰能够成功分离,但运行时间将会很长。另一方面,运行时间明显更短且分离度良好的分离可缩短运行时间,但其上样量可能较低。另一个考量因素是下游处理或馏分回收,其溶剂用量或溶剂类型也是很重要的因素。最后,如果可在等度条件下实现分离,则可采重迭进样大幅提高效率。选定色谱柱和溶剂后,可通过调节其它参数在进行放大和纯化之前进一步优化分离效果。
优化梯度条件:SFC中梯度聚焦的作用方式与RPLC有所不同。由于正相色谱中存在许多竞争保留机制,因此梯度变化对保留效果产生的影响会因分析物而异。就SFC而言,在助溶剂比例随梯度增加时,会伴随密度和压力降低的现象。助溶剂增加量与保留时间变化并非呈线性关系,因此我们难以预测不断变化的条件下化合物的选择性。对于结构相似化合物,由于其遵循相似的保留机制,该问题影响不大。不同结构的化合物组成的混合物(例如存在于基质中)则比较棘手。不过一般来说,采用平缓梯度可以延长保留时间和提高分离度。图27所示为SFC分离指定目标化合物的梯度聚焦示例。
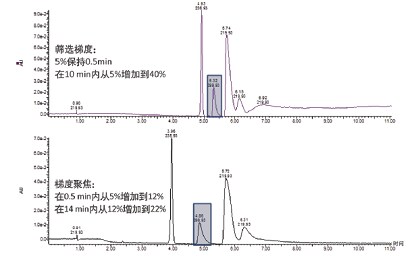
图27. 应用梯度聚焦后色谱分析结果的变化。
确定等度条件:等度方法不仅易于根据筛选结果开发,而且可执行重迭进样以提高分析效率,是一种非常理想的方法。洗脱时的助溶剂比例可通过保留时间、系统的筛选梯度斜率和补偿,以及色谱柱体积延迟来确定。对于SFC,理想的优化起始点是该比例的计算值减去5%。
以图28所示的分离为例,筛选梯度为5 min内从2%升至20%,而梯度延迟为0.46 min(预先测得)。计算可得梯度斜率为3.6%/min,起始百分比为2%,按照如下公式计算洗脱第一个峰时(4.12 min)助溶剂的比例:
洗脱时的助溶剂比例= (保留时间–梯度延迟) x 梯度斜率 + 起始比例
洗脱时的助溶剂比例= (4.12 min – 0.46 min) x 3.6%/min + 2%
洗脱时的助溶剂比例= 15%
因此,减去5%后,等度条件的助溶剂优化起始点应为10%。色谱分析结果表明分离度良好,不过将流动相中的助溶剂比例增加到15%后,色谱峰分离度仍然很出色,并且运行时间更短。因此在本例中,10%方法允许更大的上样量,而15%方法的运行时间更短。
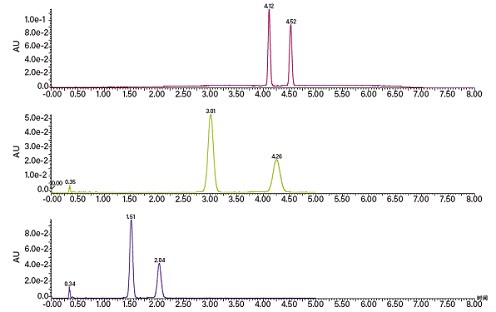
图28. 在梯度筛选条件下分离手性化合物所得的色谱图,以及基于其保留时间进行等度方法开发所得的色谱图。
如何选择制备型系统
系统规模
不同Prep SFC仪器可在低至2 mL/min到高达数百mL/min的流速下运行,能够满足不同规模的分析需求⁷。在正确的工作流程中,纯化规模必须与应用目标相匹配,这一点对于样品量有限或样品溶解度低的应有尤为重要。在这种情况下,小规模(半制备型)纯化系统更加实用。对于SFC,小规模纯化一般采用内径4.6 mm~10 mm的色谱柱,流速范围为3~20 mL/min。Prep SFC中色谱柱规格与流速和预计载样量的匹配表如下所示(表7)。需要注意的是,特定样品的载样量取决于溶解性、目标化合物的分离度及其在基质中的相对含量等多个因素。
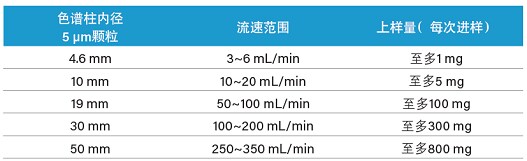
表7. 色谱柱内径、流速和每次进样的载样量估算值表。
较大规模的Prep SFC系统采用18~50 mm内径的色谱柱,流速范围为50~350 mL/min。大容量色谱柱可提高每次进样的上样量和流速,从而优化此类应用的通量。不过,在包罗万象的纯化领域,上述系统只能算中等规模的系统。现在已有许多成熟的工业工艺采用大型中试级Prep SFC系统(此处未介绍)。这些系统采用大型(内径30 cm或更大)高压动态轴向压缩(DAC)色谱柱,并且通常作为工厂设施运行。
工作流程:“大批量”还是“批次”
根据应用不同,Prep SFC可提供两种基本工作流程。第一种称为“大批量”纯化。在该工作流程中,大批量的某种样品将被多次进样,直至目标化合物达到指定回收量或样品耗尽。系统将收集所有符合条件的馏分并集中混合到一定数量的收集瓶中的一个。此法可处理“大批量”物料,并进行若干小时(有时甚至是数天)的回收。通常采用UV引导的纯化方法进行此操作,但也可采用其它检测手段。这种工作流程通常用于已对样品进行过准确表征、对其有充分了解,并且需要提高生产效率的情况。
许多“大批量”应用都采用重迭进样法。重迭进样可利用所有可用的色谱空间进行连续分离和纯化,能够显著提高通量。从本质上来讲,重迭进样可缩短进样周期之间的时间,从而进一步将溶剂用量降至尽可能低。重迭进样示例见图29。在有些应用中,可在第一组色谱峰洗脱之前多次进样。要采用重迭进样,系统必须能够在收集馏分的同时执行进样。因此,需使用单独的进样模块,或者能够自主移动收集和进样组件(进样针和探头)的系统。
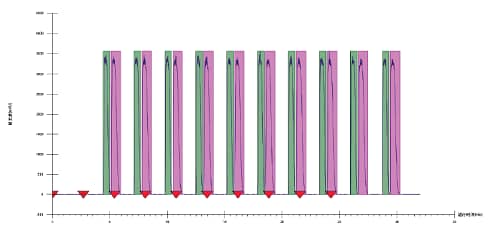
图29:氟比洛芬酯对映体的重迭进样和收集。
“批次”纯化是十分实用的实验室纯化工作流程,还适用于需要纯化多个样品中的多种目标化合物的情况。通常采用质谱引导的纯化,因为该方法在收集馏分时选择性更高。如果采用光学检测(或UV)引导的纯化,由于许多化合物的吸收波长相同,检测器将无法区分色谱峰。质谱引导的纯化基于化合物质量数收集馏分,该参数可有效区分目标化合物与任何杂质,因此更具针对性。当样品基质较为复杂、样品尚未经过准确表征,或者样品因不具有发色团而无法采用UV检测时,质谱引导的纯化方法尤其有用。通常采用梯度方法来改善峰形以及更好地分离化合物与复杂干扰物。该工作流程采用开床式收集,将馏分收集到多个试管中,通常每根试管仅含一种馏分。样品按“批次”运行,系统根据质量数收集目标化合物。批次纯化也可仅由UV触发,或者由UV和MS共同触发。图30所示为采用MS触发方法纯化天然产物中的目标化合物的示例。
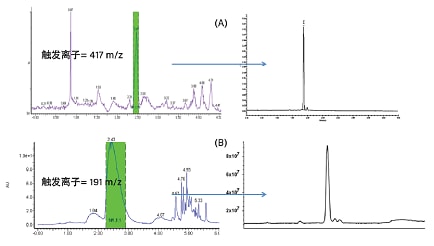
图30. 采用MS触发方法纯化天然产物中的目标化合物,样品为:(A)五味子提取物和(B)当归根部提取物18,19。
