质谱引导的纯化注意事项
质谱引导的纯化注意事项
传统的肽分离通常采用UV检测法,而质谱引导的分离方法可以更加明确地区分目标肽与合成及裂解过程中形成的污染物,从而使纯化过程变得更加轻松。质谱检测可用于评估复杂色谱图中色谱峰的鉴定结果和纯度,同时提高样品通量。开发应用质谱和UV检测技术的分离方法有助于我们获得更完整的样品色谱图。无法电离或难电离的化合物通常采用短波长UV检测。相反,UV吸光性极差的肽通常可由MS轻松检出。
溶剂
肽分离常用的溶剂(包括乙腈、甲醇、乙醇和异丙醇)均可兼容电喷雾电离(ESI)和其它大气压电离技术。不过,通常使用乙腈时的色谱分离度、选择性、峰对称性和分析效率最佳。由于粘度较低,采用乙腈进行色谱分离还能降低LC系统背压。溶剂的挥发性和提供质子的能力对于ESI也非常重要。挥发性有机溶剂可降低MS源内形成的液滴的表面张力,有助于提高电离效率和分析灵敏度。许多肽的分析和分离过程都会用到酸性改性剂(例如甲酸或三氟乙酸),尽管三氟乙酸对电离有一定程度的抑制作用。如果方法开发过程涉及正负离子切换,可兼容MS的醋酸铵或甲酸铵流动相是可接受的选择。对于在高pH下分离效果更好的肽,适合采用含碳酸氢铵的流动相。为了避免样品在质谱检测器中沉淀,应采用浓度小于等于20 mM的挥发性缓冲液。
检测控制
进行制备型色谱分离时必须特别注意检测环节。高浓度组分的峰可能会超出大部分检测器的线性范围,此外,检测器硬件无法兼容过高的流速。某些检测器(如MS和ELSD)属于破坏性检测器,因此有必要适当降低样品进入检测器时的流速和浓度,同时尽可能地减少样品损失。通常使用被动分流器在样品到达检测器前对其进行分流和稀释(图47和图48)。补偿溶剂可稀释制备级流量采样,并将其输送至检测器。被动分流器专用于特定的色谱分析流速范围和分流比。分流器流路必须与适用于选定色谱柱的流速相匹配。若色谱峰在较高浓度处洗脱,应采用较大的分流比。常用分流器及其分流比如表6所示。

图47:被动分流器。
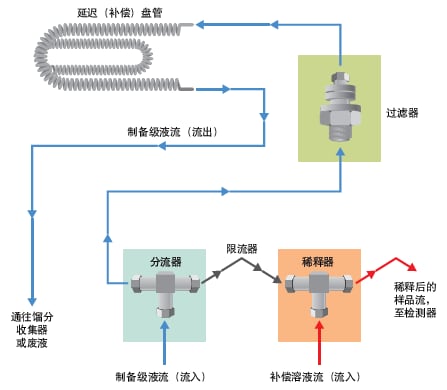
图48:被动分流器液流示意图。
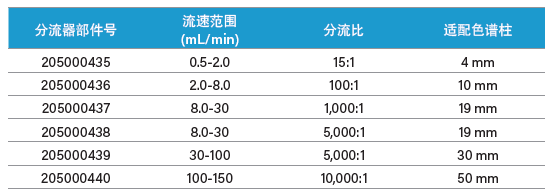
表6:沃特世被动分流器。
作为被动分流器的替代装置,主动分流器按照与设定分流比对应的速率对制备级流量进行机械采样,然后由补偿溶剂对采集的样品流进行稀释并输送至检测器。然而,主动分流器常常会干扰基线,此外机械磨损也会时常影响装置性能。被动和主动分流器在制备级流量采样方面的性能相当,因此具体采用哪种类型的分流器取决于用户偏好。
改性剂
具体选择哪种流动相和改性剂取决于分离得到的肽将用于何种用途。如前所述,流动相改性剂是一种添加剂,其通过升高或降低流动相pH来控制肽的离子特性。改性剂还可与肽的带电侧链和末端形成离子对,从而增强样品的疏水性。一般使用三氟乙酸达到上述目的。离子对的形成可促进肽与疏水性色谱柱固定相之间的相互作用,进而改善分离质量。然而,肽与TFA形成的强离子对难以在电喷雾电离条件下分离,导致离子化的肽分子数减少并削弱信号强度。理想的改性剂应该既能通过形成离子对改善色谱分离,又不妨碍电喷雾电离。
Apffel等人提出通过柱后添加弱酸的方法与TFA竞争配对分析物。浓度足够高的弱酸可促使竞争配对向TFA质子化的方向进行,质子化的TFA可通过蒸发去除,而让分析物实现更加高效的电离。这有助于提升MS信号的灵敏度和强度6。为此,可选用多种不同浓度的弱酸,例如,在质谱引导的纯化中使用含0.1%丙酸的1:1水/有机溶剂溶液作为补偿溶剂可成功达到上述效果。
单同位素质量数与平均质量数
质谱引导的纯化可降低目标肽分离的不确定性,而选择合适的质量数用于馏分触发非常重要。大多数小分子分离主要采用单同位素质量数进行鉴定,其它的相关离子加合物则根据该值来确定和进行监测。对于肽等分子量较大的分子,有时需要使用平均质量数作为主要鉴定参数。
单同位素质量数是采用分子中各元素丰度最高的同位素的质量数计算得出的,而平均质量数是采用分子中各元素所有天然同位素的加权平均数计算得出的。因此,肽的平均质量数明显大于其单同位素质量数。随着分子中的碳原子数增加,出现13C的可能性也越大,分子量也会随之增大。此外,单同位素质量数可能不是质谱图中丰度最高的峰。虽然关于使用单同位素质量数还是平均质量数并没有硬性规定,但一般来说1500~1700的分子量范围是改用平均质量数触发肽馏分收集的分界点。在实际应用中,市面上的许多软件程序都会同时计算各种肽电荷态的平均和单同位素m/z。十分谨慎的做法是制定两种质量数触发方案,通过分离之前的筛查运行,将很容易确定理想触发方案。
电荷态
电喷雾电离技术在温和条件下电离样品,并且可生成带多个电荷的分子,是质谱引导的纯化常用的一种电离技术。尽管某些肽的分子量已经超出了分析仪的质量数上限,但多电荷离子的m/z值较低,一般都在质谱仪的质量数范围之内。溶液pH、酸性和碱性官能团数量以及分析所用溶剂的物理性质等诸多因素都会影响肽的电荷态。
如前文所述,肽分离的第一步通常是分析。计算出分子量和可能的电荷态,便于肽产物的鉴定。粗制样品分析不仅有助于我们确定需要实施何种水平的方法开发才能达到可分离出纯产物的足够分离度,还能提供有关肽电荷态的信息。以来自粗制样品分析级分析的总离子流色谱图对应的质谱图为例(图49),计算所得的肽质量数为1772.9,存在带正电的离子(1773.9),但数量极少,而目前质量数为887.8的双电荷离子丰度最高。图中还存在少量三电荷离子(592.3)和杂质(781.2)。尽管质谱图中存在多种电荷态的离子,但目标肽在色谱图中仅表现为单峰。在提取离子流图中(图50),和我们预期的一样,双电和和三电荷离子峰同时洗脱。
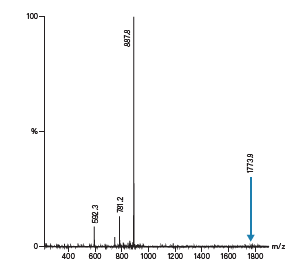
图49:来自粗制肽分析级分析的总离子流色谱图(TIC)的质谱图。
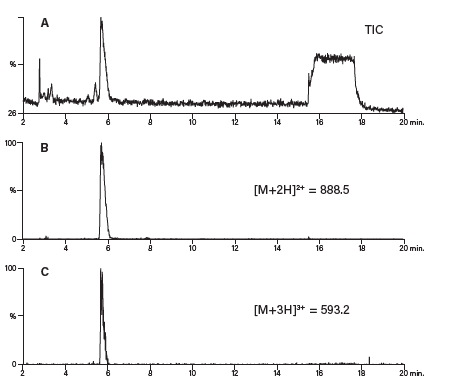
图50:多电荷离子的洗脱。
馏分收集触发器
肽带有多个电荷常常会导致我们难以预测给定MS实验中丰度最高的离子。因此,采用质谱引导的纯化方法进行肽分离时,馏分触发器必须涵盖所有可预见的多电荷离子。
数据采集:连续模式与质心模式
鉴于肽样品较为复杂、可能存在多种电荷态,而且随着肽链长度增加可能需要由不同的质量参数(单同位素质量数或平均质量数)来触发馏分收集,应采用连续模式采集MS数据。连续数据可显示质量数轴上所有分离点的实测强度,它包含了测定特定强度的m/z时引入的统计不精确度。我们随后可通过处理这个原始信号得到各个可能发生重叠的质谱信号的强度37,从而区分邻近的相似m/z。质心数据是经过处理的连续数据,其显示质谱图中每种离子分布的单一中心点,并且在质谱图中以棒状图的形式显示(图51)。
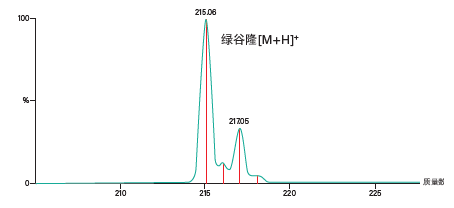
图51:连续质谱数据与质心质谱数据的叠加图。
质谱引导的肽分离示例
按照上述原则,采用质谱引导的纯化方法分离两种肽(由Research Genetics公司的KellyWasmund博士提供,公司位于美国阿拉巴马州亨茨维尔):

分别用0.5 mL二甲基甲酰胺(DMF)溶解两种肽,然后用水稀释至5.0 mL。ISQA的估算浓度为5~10 mg/mL,SIIN的估算浓度为10 mg/mL。对两种肽进行小规模分离的条件见表7;图52和图53分别为色谱和质谱分析结果。ISQA和SIIN各电荷态目标离子的计算结果如表8所示。
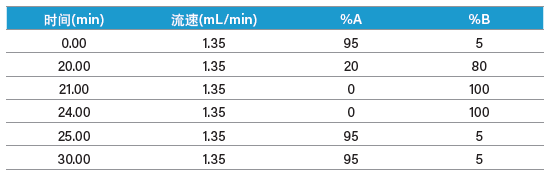
表7:ISQA和SIIN小规模分离的梯度条件。
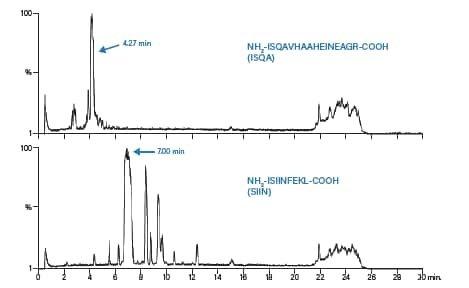
图52:ISQA和SIIN小规模分离的总离子流色谱图。
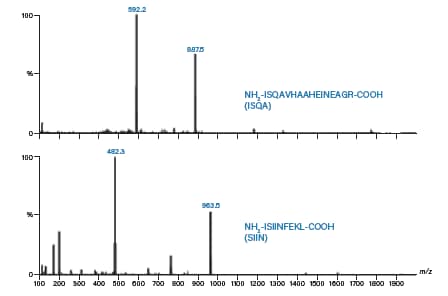
图53:ISQA和SIIN的质谱图。
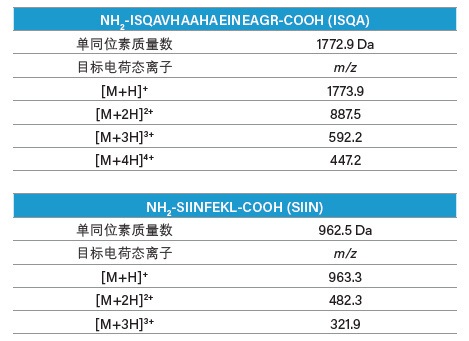
表8:ISQA和SIIN各电荷态目标离子的计算结果。
我们分别为两种肽制定了梯度聚焦,以提高目标肽与其邻近洗脱污染物之间的分离度。ISQA制备型分离的梯度聚焦从12% B开始增加至20% B,目标肽在17%B附近洗脱(表9和图54)。类似地,SIIN制备型分离的梯度聚焦从23% B开始增加至31% B,目标肽在28% B附近洗脱(表10和图55)。用于馏分收集的质量触发器包括可预见的多电荷离子(表8)。
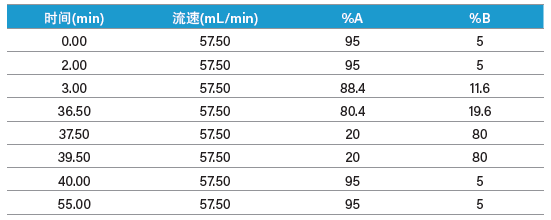
表9:纯化ISQA采用的制备级梯度聚焦。
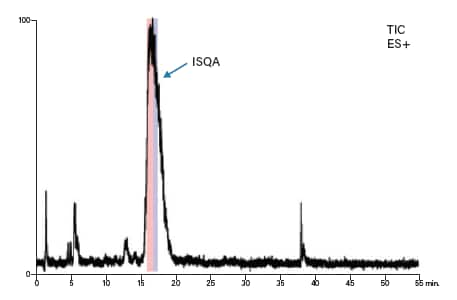
图54:ISQA的制备级MS谱图。
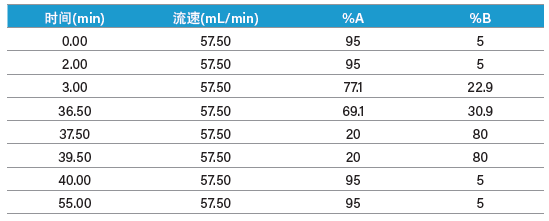
表10:纯化SIIN采用的制备级梯度聚焦。
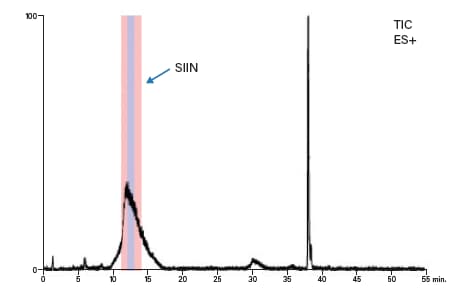
图55:SIIN的制备级MS色谱图。
对制备型运行得到的馏分进行分析,以评估其纯度。采用多个检测通道监测分析结果,从而对馏分进行全面表征。ISQA和SIIN肽的馏分分析均采用与粗制肽分析相同的梯度方法。ISQA和SIIN的分析结果分别为图56和图57。
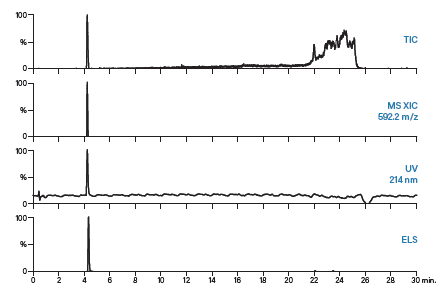
图56:ISQA的馏分分析结果。
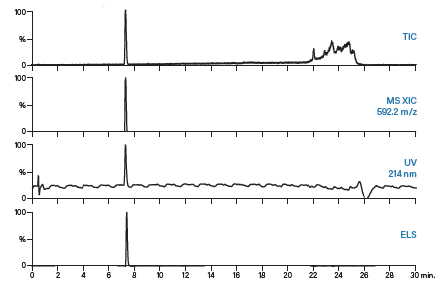
图57:SIIN的馏分分析结果。
结语
随着合成、药物设计、发现、开发和生产方面的技术不断更新和改进,肽类药物正逐渐填补治疗药物领域的重要技术空白。尽管技术不断革新,肽分离的基础始终是基于反相HPLC与UV和质谱检测联用的各种分离方案。先采用快速梯度分析粗制肽,然后设置梯度聚焦并将分离方法几何放大至配备柱头稀释和温度控制装置的大规格色谱柱上,是快速分离目标肽的有效途径。尽管肽分离广泛采用UV检测,但质谱引导的纯化可减少收集到馏分数量,从而缩短后续馏分分析所需的时间。利用分流和稀释技术控制检测器信号、使用合适的色谱流动相、改性剂和补偿溶剂以及合理选择触发馏分收集的电荷态均有助于简化肽纯化方案。
