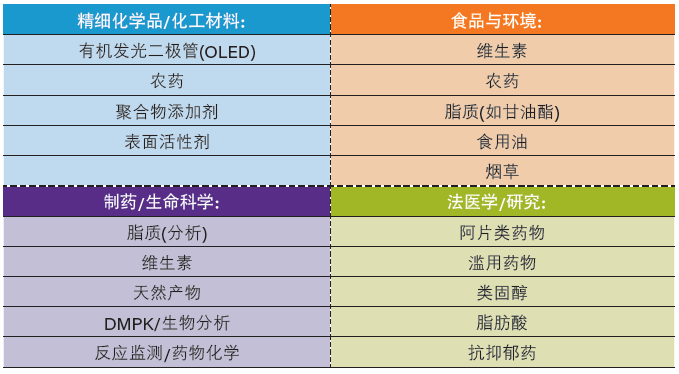
合相色谱应用范围
CC应用范围
如前文所述,合相色谱的选择性非常广泛,适用于多种应用(表6)。
无论在哪个市场领域,也无论目标分析物的具体性质如何,CC都能从以下三个重要方面协助分析人员应对分析挑战:
- 合相色谱可简化工作流程
- 合相色谱可分离结构相似化合物
- 合相色谱是反相液相LC的正交分离模式
接下来的内容将通过一些应用示例详细介绍CC的主要优势。
表6:按市场领域和化合物类型分类的CC应用
简便性
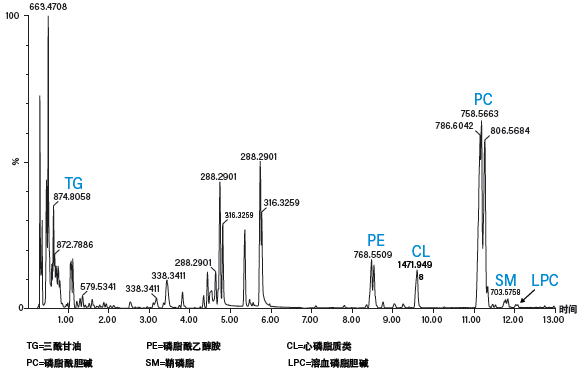
图42:使用配备MS检测器的ACQUITY UPC2系统分析小鼠心脏提取物得到的脂质综合分析谱图。
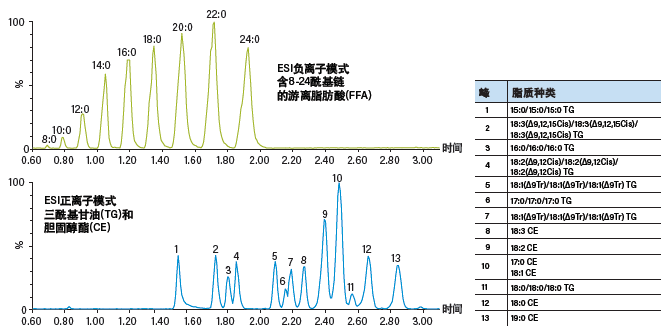
图43:使用配备MS检测器的ACQUITY UPC2系统对游离脂肪酸(FFA)、三酰甘油(TG)和胆固醇酯(CE)进行靶向分析的结果。
在SFC到CC的演进过程中,尤为有用的发现之一是压缩CO2可与各类有机溶剂混合,因此我们能够以优秀的方式进行色谱分析。本节将介绍CC如何大幅简化分析实验的工作流程。
对于所有分析实验室而言,从最初的样品采集到最后的分析,任何环节的工作流程简化都会对业务产生巨大影响。CC能够大幅简化许多应用的工作流程,有效节省成本和时间、降低出错率并提高工作效率。上述简化体现在以下方面:
- 结合多项技术(LC和GC)
- 结合多种方法(正相LC和反相LC)
- 缩短样品制备时间
结合多项技术 — 脂质分析
出于诸多原因,脂质分析意义重大。在制药行业中,人们借助脂质分析来确定对照和受试者体内的药效影响。
在临床研究中,脂质可作为不同疾病的生物标志物,还可衡量治疗的疗效。食品应用则通过分析特定种类的脂质(如甘油三酸酯)来评价食物营养水平或辨别产品真伪。化工原料行业会分析石油产品(如生物柴油)中的脂肪酸和甘油三酸酯。根据预期目标,脂质分析需要采用不同的技术。人们通常采用GC来分析游离脂肪酸,使用该方法时需要将游离脂肪酸(尤其是长碳链脂肪酸)衍生为脂肪酸甲酯(FAME)以改善峰形和检测限。衍生化步骤需要花费数小时,而且随后的GC分析耗时长达30 min。在分析磷脂和鞘脂类等强极性脂质时,通常需使用HILIC或正相LC来分离不同种类的脂质(基于其极性头部基团的性质)。然后,根据碳链长度和/或双键数量的差异,使用反相LC分析同种类的强疏水性脂质。由此可见,全面的脂质分析要用到多项技术。但使用CC时情况并非如此,它只需一次进样即可分离所有种类的脂质。图42所示为采用ACQUITY UPC2系统分析小鼠心脏提取物得到的脂质综合分析谱图。在本例中,我们使用BEH色谱柱和通用梯度成功分离了不同种类的脂质,所得分离结果与正相LC或HILIC的结果相当。
对于中性脂质(如三酰甘油(TG)、甘油二酯(DG)、胆固醇酯(CE)以及游离脂肪酸),只需改变色谱柱和梯度条件即可根据脂肪酸碳链长度以及双键数量的差异保留和分离同一种类中的不同脂质(图43)。
在本应用中,CC不仅比GC分析速度更快(速度提升多达10倍),而且无需衍生化步骤,因此可大幅简化整个脂质分析工作流程。省去衍生化步骤可缩短分析时间,还能尽可能减少在工作流程中增加这些额外步骤时可能引入的错误。如果不使用CC,此类分析和靶向分析需要使用多达三种不同的技术,导致样品通量低、溶剂用量大、运行时间长,整体分析成本也更高。
结合多种LC方法 - 脂溶性维生素
脂溶性维生素分析在制药行业、临床研究和诊断,以及食品和燃料行业中具有十分重要的意义。人们通常使用反相或正相LC分析脂溶性维生素和类胡萝卜素(表7)。由于这些化合物难以通过单次进样实现分离,一般需要使用不同的色谱柱和流动相对它们进行单独分析,分析时间为1030 min。相比而言,CC在10 min之内即可完成所有维生素和相关化合物的分析(图44)。与表7中所列的传统分析方法不同,经简化的CC方法只需使用一种色谱柱、一种流动相条件以及一种检测器。CC可直接进样提取或溶解目标化合物时常用的溶剂(例如异辛烷和正己烷),而无需进行反相分析中通常必须执行的溶剂交换步骤。
表7:脂溶性维生素及相关非极性化合物的典型分析条件
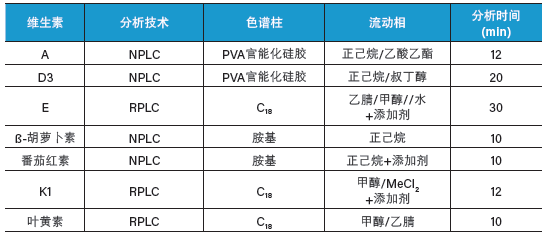
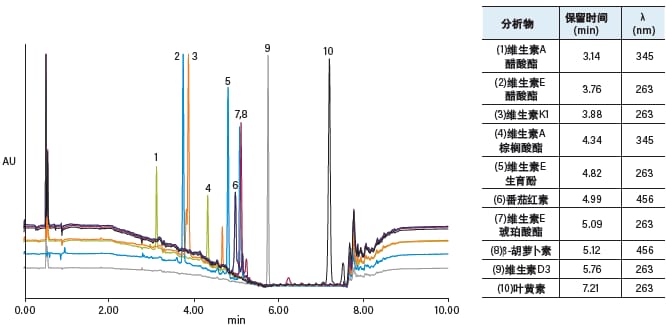
图44:使用单一CC分析方法分析10种维生素标准品的叠加谱图
结合多种LC方法 — 杂质分析
分析复杂样品时,由于样品中特定组分的化学和物理性质各不相同,方法开发的难度往往比较大。正因如此,拥有多种可应用于方法开发的工具(如不同模式的色谱分析工具)对于科学家而言将会很有帮助。例如,将传统液相色谱与质谱联用,可以开发出活性药物成分(API)昂丹司琼及其相关杂质(图45)的高灵敏度分析方法。
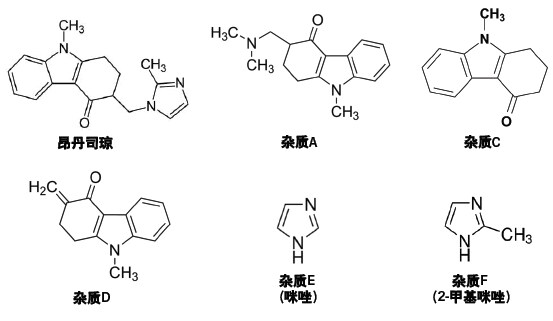
图45:昂丹司琼及其相关杂质的结构
初步的探索运行结果表明,典型反相条件无法保留小分子极性杂质咪唑和2-甲基咪唑(图46)。RPLC在不使用离子对试剂时对极性化合物的保留性很差,这是该技术的一个已知缺陷,而采用质谱检测时往往不会使用离子对试剂,因为它们会抑制MS信号。使用正交液相色谱方法(具体而言即亲水作用色谱(HILIC))有利于以上两种极性化合物的保留。但是,由于化学性质的差异,HILIC方法无法保留杂质C和D(图47)。在本例中,我们采用两种液相色谱法来分析昂丹司琼及其相关杂质:采用反相方法分析杂质C和D,采用HILIC方法分析杂质E和F。使用这两种方法定量分析浓度在15-500 ng/mL范围内的五种杂质,六次重复进样具有良好的准确度和精度(< 15%),并且所有化合物的校准曲线R2值都> 0.995,表明该方法的稳定性优秀。
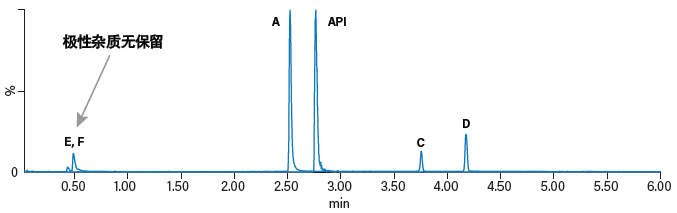
图46:在通用反相LC条件下分离昂丹司琼(API)及其相关杂质A、C、D、E和F的色谱图。使用ACQUITY HSS T3色谱柱(2.1 x 100 mm, 1.7 μm),流动相为0.1%甲酸水溶液和0.1%甲酸的乙腈溶液。
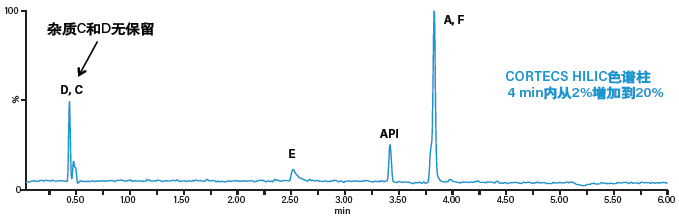
图47:在HILIC条件下分离昂丹司琼(API)及其相关杂质A、C、D、E和F的色谱图。使用CORTECS HILIC色谱柱(2.1 x 100 mm, 1.6 μm),流动相为0.1%乙酸水溶液和pH4的10 mM乙酸铵水溶液。
合相色谱拥有与正相色谱十分相似的保留机制,同时又使用完全与质谱兼容的流动相,因此是解决此类分析难题的理想替代方法。采用基于UPC2 Torus色谱柱的方法开发策略,我们开发出了使用Torus 2-PIC色谱柱通过单一方法分离并定量昂丹司琼及其五种相关杂质的分析方法(图48)。
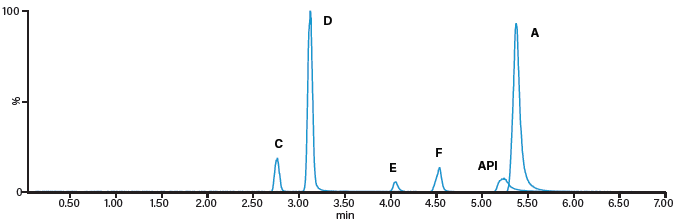
图48:使用ACQUITY UPC2分离昂丹司琼(API)及其相关杂质A、C、D、E和F的色谱图。使用Torus 2-PIC色谱柱(3 x 100 mm. 1.7 μm),以0.2% NH4OH的甲醇溶液作为助溶剂和补偿溶液流。
采用这种合相色谱(CC)方法定量分析浓度在相同范围内(15-500 ng/mL)的五种杂质,六次重复进样同样显示出良好的准确度和精度(< 15%),并且所有化合物的校准曲线R2值都> 0.995。我们开发的CC和LC方法在线性、重现性和检出限方面性能相当,均适用于常规分析。不过,LC方法要使用2根色谱柱、2组流动相和2种进样方法,而CC方法仅需使用1根色谱柱、1组流动相和1种进样方法,可以有效节省时间、资源和消耗品。由此可见,对于任何方法开发实验室而言,引进CC技术都将大有裨益。
缩短样品制备时间
除了可将多种技术和方法相结合,以更快的速度分离分析物之外,CC通常还能缩短样品制备时间。CC兼容有机溶剂,因此拥有以下优势:
- 无需水解和/或衍生化步骤
- 无需通过蒸发和复溶进行溶剂交换
- 操作步骤少,可有效减少实验误差和提高数据质量
以分析食品中的脂溶性维生素为例,需要执行多个样品制备步骤和多次分析。图49所示为食品中维生素A、D和E的典型样品工作流程。由图可见,维生素A和E的样品制备步骤与维生素D不同。另外,需要对这三种维生素进行三次独立的HPLC(正相和反相)分析。维生素D的样品制备过程尤其复杂,其包含多个步骤,在某些情况下还包括半制备型HPLC分离步骤。
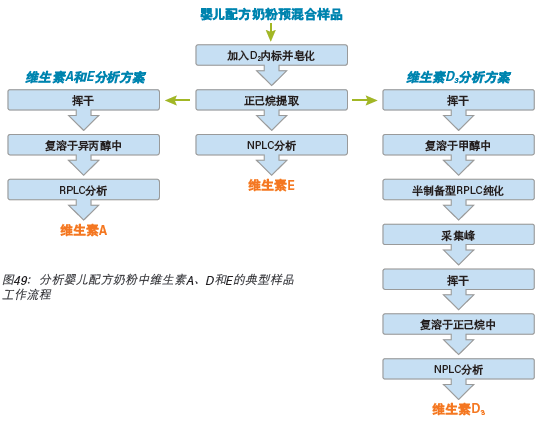
图49:分析婴儿配方奶粉中维生素A、D和E的典型样品工作流程
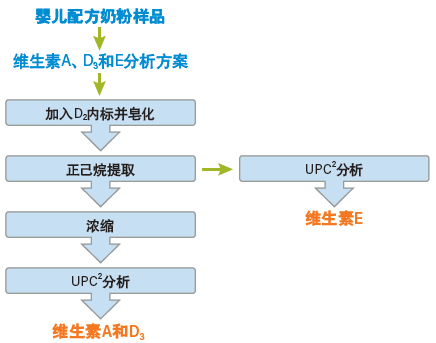
图50:使用CC分析婴儿配方奶粉中维生素A、D和E的样品工作流程
使用CC分析相同维生素的样品制备步骤要简单得多(图50),因为该技术兼容提取过程前期使用的非极性有机溶剂。在本例中,我们直接进样经己烷提取的样品即可定量分析维生素E,然后对样品进行浓缩之后分析维生素A和D3,这样就使得运行时间比传统分析方法缩短了20倍。另外,CC只需三个样品制备步骤、一种方法以及一台仪器,而图49所示的传统工作流程包括12个样品制备步骤、三种方法,还需使用两台不同的仪器。表8总结了CC用于上述应用的优势,并列出了该技术还可在哪些其它领域让分析科学家受益于如上所述的精简分析流程。
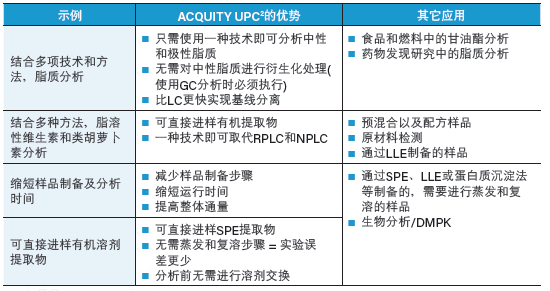
表8:使用ACQUITY UPC2系统简化工作流程的优势
快速分离结构相似化合物
由于具有结构相似性,异构体和结构类似物(尤其是光学异构体)有时很难实现分离。接下来我们将讨论CC在分离以下结构相似化合物中的应用:
- 手性分离(对映体和非对映体)
- 位置异构体(官能团位置不同)
- 结构类似物
——生物标志物(共轭/非共轭)
—— 药物(代谢物、杂质、降解产物)
手性分离
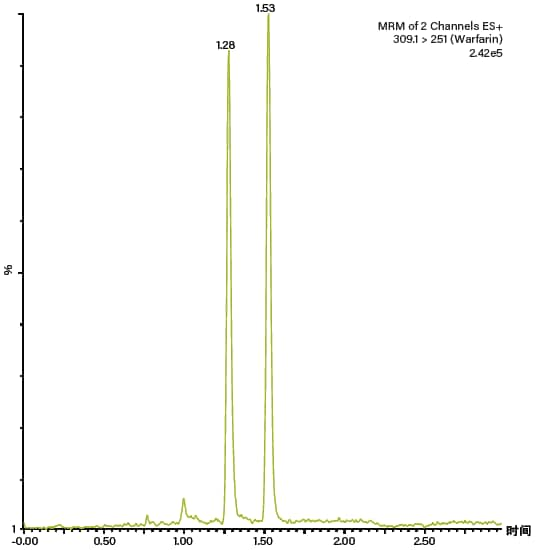
图51:使用CC分离血浆中的华法林对映体
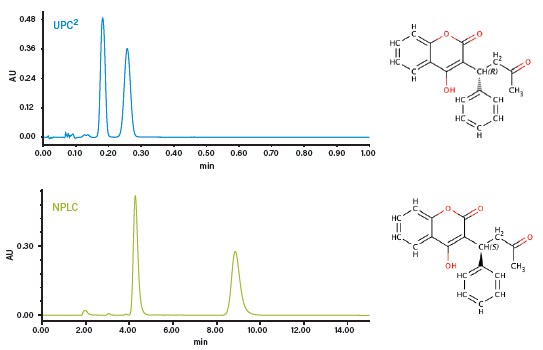
图52:使用CC和正相LC分离华法林对映体
化合物的不同对映体通常具有不同的功效和毒性特征,因此在研究、开发和生产阶段须对其进行监测。人们主要使用纤维素或直链淀粉基固定相通过正相LC进行手性分离。在正相LC中,梯度分离难以实施。分析人员必须使用不同的流动相组合(通常需要毒性很强的溶剂)和不同的色谱柱进行多次等度分析,因此方法开发过程相当耗时。CC能够运行梯度分析,而且选择性范围极广,只需使用无毒的溶剂,借助该技术,分析科学家们在一天之内即可开发出手性分离方法。
多年以前,纯化化学家们就已经认识到了SFC在手性分离中的应用价值。尽管分析型SFC分离的准确度欠佳,但对于快速手性筛选、手性方法开发、对映体过量转化(e.e值)测定和手性转化研究而言是非常理想的。与正相LC不同,CC与质谱检测高度兼容,因此能够鉴定并表征反应、生产过程以及生物系统中的对映体及其构型(图51)。
图52对比了华法林对映体在正相和合相色谱上的分离情况。与正相色谱相比,CC在极短的时间内就实现了对映体的基线分离(比正相色谱快30倍)。另外,由于无需使用购买和处理成本都很高的有毒溶剂,CC将每次手性分离的分析成本降低了多达100倍。所有这些优势使得CC成为了各种手性分析的理想技术。
位置异构体和结构类似物
CC在位置异构体和其它结构类似物的分离中非常有用。位置异构体是分子量相同(同量异位)但官能团位置不同的一类化合物。涉及原料分析、反应监测以及不对称催化的应用中常常出现这类化合物。为了分离异构体,这些化合物在GC分析前一般都需要进行衍生化。正相LC方法有稳定性欠佳、分析速度慢的固有缺陷。另一方面,具有选择性优势的CC分离方法在通用条件下就能轻松分离位置异构体,且无需衍生化步骤。
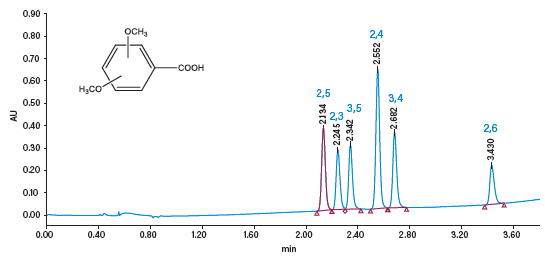
图53:使用CC分离二甲氧基苯甲酸(DMBA)位置异构体
ACQUITY UPC2系统(图53)可快速分离异构体,有助于实时评估反应原料、中间体和最终产品的优化效果。结构彼此相似的结构类似物很难实现分离,此类物质包括共轭或非共轭的生物标志物(如葡糖苷酸、硫酸盐)以及代谢物、降解产物和药物化合物的杂质。类固醇是最常见的一类结构类似物(图54)。不同类固醇的结构相似,由于其质量数差异非常小,即使采用MS检测也很难对它们进行分离和分析。采用CC,在多种色谱柱上应用通用筛查梯度都能在2 min内轻松分离这些化合物(图55)。由于此类化合物的非极性特点,此类分离对反相LC而言非常棘手,而GC方法则需要进行衍生化以改善峰形和检测限。ACQUITY UPC2系统与MS检测联用是鉴定和定量类固醇的一种优良方法。
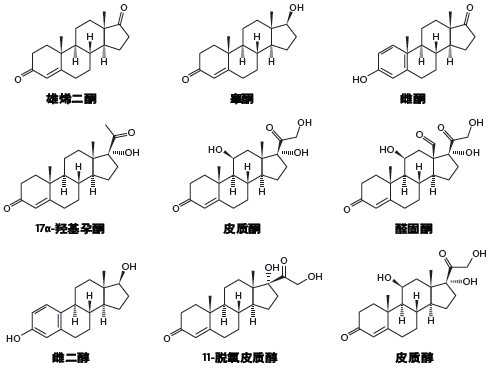
图54:非共轭(游离)类固醇的结构

图55:使用合相色谱分离九种类固醇
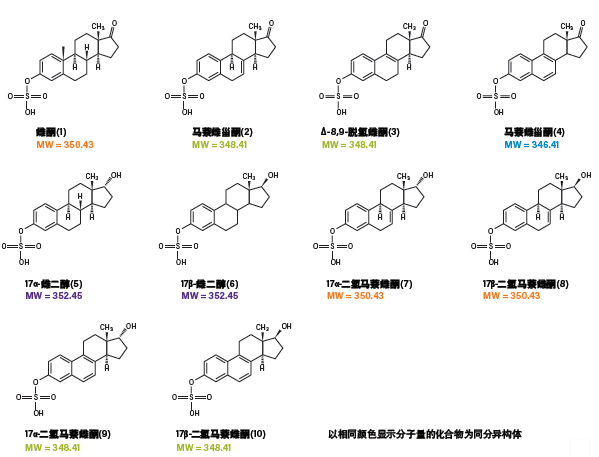
图56:硫酸化雌激素的结构,颜色相同的分子量表示互为同分异构体
分离共轭结构类似物的难度更大。游离类固醇(图54)不溶于水,因此人体会将它们转化成硫酸化的水溶性衍生物。这个过程会生成带负电荷的亲水性侧基,使它们可溶于水(图56)。人们从天然原料中分离出这类化合物用于疾病治疗,在疾病研究和疗效评估中,它们被用作生物标志物。这些化合物的分析面临两个主要挑战。
首先,GC分析耗时30 min,而且样品制备过程包括硫酸基团的酶水解及后续的衍生化步骤,耗时2.5 h。其次,由于某些雌激素是同分异构体(m/z相同),质谱无法区分它们。因此,需要借助色谱法分离不同形式的同分异构体。
CC可在15 min内分离所有10种硫酸化雌激素(图57),其中包括两个邻近洗脱峰(峰6和7),而采用30 min的GC分析方法时,这两个峰难以分离(图58)。由于CC能够分析硫酸化化合物的原本形式,因而样品无需进行水解和衍生化。这样一来大大减少治疗制剂的分析步骤,因此通量和效率得以提升。
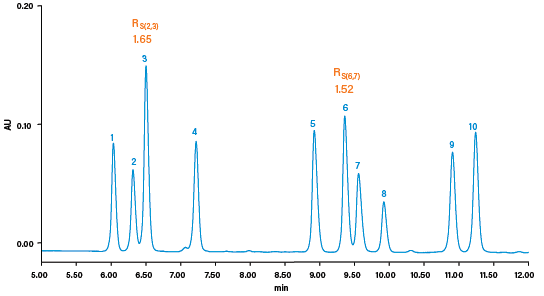
图57:使用CC分离10种硫酸化雌激素的结果
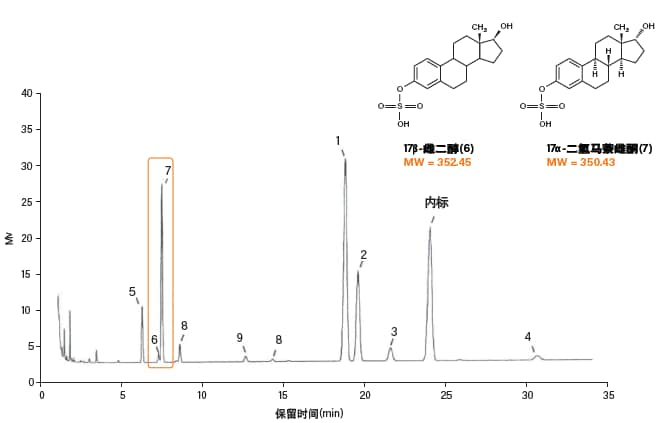
图58:采用共轭雌激素的USP分离方法,通过GC-FID分离10种雌激素的结果。制备样品时,需要在对样品进行化学衍生化处理之前从共轭雌激素上裂解硫酸基团。整个样品制备过程耗时2.5 h以上。其中两种化合物(峰6和7,用红框圈出)没有完全分离开。
表9:使用CC分离结构相似化合物的优势
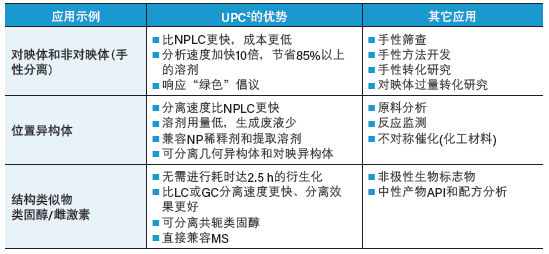
表9总结了CC用于上述应用时的优势,同时给出了适合采用该技术分离对映体、位置异构体以及结构类似物的其它应用领域。
正交性
正交分离模式彼此互补,但各自又以其独有的方式实现峰保留,因此获得的样品信息比单一分离模式更丰富。拥有采用不同技术分离分析物的能力极其重要,原因如下:
- 有助于可靠地鉴定和表征杂质、降解峰或相似化合物(例如同分异构体或共洗脱化
合物)
- 确保样品得到全面表征
- 能够获得更丰富的样品信息
- 有助于从基质干扰中分离目标化合物
正交分离技术的例子包括互补模式,例如正相和反相色谱。CC的选择性与正相色谱类似,但它比任何正相方法都更加稳定可靠(请参阅第2章),而且重现性更好。接下来的内容将展示以CC作为正交分离模式实现上述目标的示例。
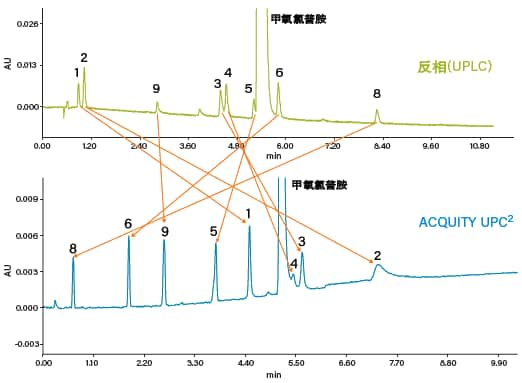
图59:以使用ACQUITY UPLC和ACQUITY UPC2系统分离甲氧氯普胺及其相关物质为例,证明合相色谱的正交性。
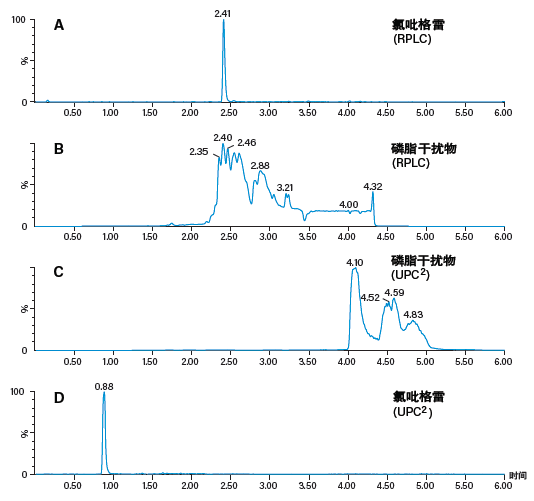
图60:使用RPLC和配备MS检测器的ACQUITY UPC2系统(MRM模式)分析经蛋白质沉淀处理的人血浆中的氯吡格雷。
使用ACQUITY UPLC和ACQUITY UPC2系统分离活性药物成分(甲氧氯普胺)及其相关物质所得的结果证明了合相色谱的正交性(图59)。采用某种技术无法分离的峰可以借助另一种技术实现分离,反之亦然。CC对极性化合物(例如峰1和2)的保留时间比RPLC更长。在本例中,ACQUITY UPC2系统成功分离了关键分析物对(峰5和甲氧氯普胺),这有助于我们进行更大规模的纯化和分离,用于后续的鉴定和表征。所有这些优势都使得CC成为了与其它常规技术配合,用于解决各种分离难题的理想技术。
在分离目标分离物与基质干扰时(例如在生物分析或食品分析中),正交方法同样发挥着重要作用。
图60A是一个典型示例,展示了使用蛋白质沉淀法从人血浆中提取的氯吡格雷的LC-MS/MS谱图。氯吡格雷具有疏水性,因此在分析中的洗脱时间非常靠后。干扰物磷脂(具有含胆碱的头部基团)也在相同的时间区域内洗脱(图60B),可能会对氯吡格雷峰造成离子抑制并导致定量结果不一致。有意思的是,ACQUITY UPC2系统洗脱磷脂干扰物时间区域与RPLC大致相同(图60C)。得益于CC与反相LC的正交性,目标分析物的洗脱时间大幅提前,与这些磷脂干扰物分离(图60D),从而尽可能降低了基质效应,确保定量结果更加准确精密。
表10总结了CC用于上述应用的优势,同时给出了分析科学家们可受益于正交分离技术的应用。
表10:CC作为正交分离模式的优势
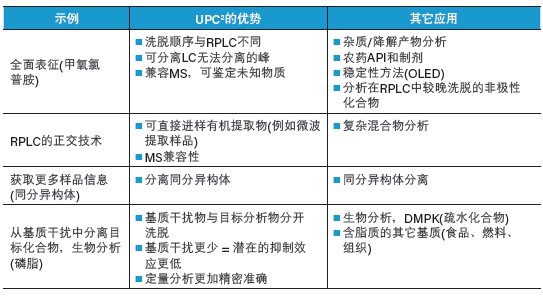
这些优势都证明了本章前面部分所提到的CC的三个重要特性:
1. 合相色谱可简化工作流程
- 集多项技术于一体
- 缩短样品制备及分析时间
- 可直接进样有机溶剂/提取物
2. 合相色谱可分离结构相似化合物
3. 合相色谱是反相LC的正交技术
- 更可靠地鉴定杂质/降解产物
- 全面表征样品
- 分离基质干扰与目标分析物