PREP SFC方法放大原则
分析级方法注意事项
将分析级方法放大为纯化方法时,确保已获得理想色谱结果非常重要。分析级方法的分离度越好,制备级色谱的结果就越好,有助于实现更高的通量和提高馏分纯度。扩大分析级方法时还应考虑制备技术条件,例如色谱柱加热、进样模式、压降、系统压力和适用色谱柱。如果采用MS引导的纯化,则分析级的所有筛选和方法开发也应采用MS检测完成。对于梯度方法,须考虑梯度斜率和平衡时间,解决分析型和制备型系统之间的体积差问题。此外还应考虑稀释剂效应,原因是分析级方法常采用混合流进样,而制备型方法采用改性剂流进样。
上样
分析人员应始终牢记不同进样策略的差异(在第3章中介绍),通常需要先进行小规模(分析型)上样研究。将样品引入系统之前,应先掌握样品中各化合物的溶解性,这一点非常重要。制备型应用的理想实践是能够使用尽可能少的溶剂上样尽可能多的样品(高浓度样品)。此外,还须保证样品被引入CO2和有机溶剂组成的流动相之后仍保持溶解状态。确定可接受的样品浓度后即可进行上样研究,在研究中逐步提高进样体积直至分离度有所损失,或者直至当前色谱分析方法无法再得到目标化合物的纯馏分。
图31所示为采用优化等度方法的上样研究示例。在本例中,进样体积超过200 μL后出现分离度损失,因此在纯化或为放大后的分离方法计算上样量时将使用200 μL的进样体积。
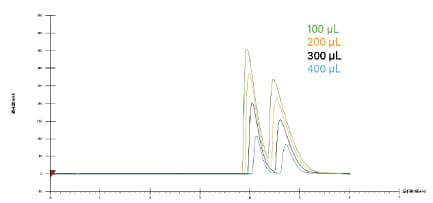
图31. 等度条件下布西丁对映体手性分离方法的上样研究。
成功放大的一般要求
开发出可接受的分析级方法并完成上样研究之后,最好在放大方法时保持保留时间和选择性不变(或相近)⁹。要成功放大色谱分析方法,以下参数必须保持一致:
流动相必须完全相同;对SFC来说,这意味着采用相同的助溶剂(溶剂B),并且CO2的质量、入口压力、物理状态(液态或气态)以及输送方式应相近。
色谱柱填料、柱长和粒径须相同,尽可能使不同规模放大方法的色谱分析结果相匹配。如果分析级方法使用小粒径色谱柱,则方法放大时色谱柱柱长与粒径的比值(L/dp)必须保持不变。
样品浓度应相同,并使用相同的稀释剂溶解。
几何放大
Prep SFC与LC相同,在放大方法时要对进样体积(上样量)和流速进行几何放大。
为了保持峰形和载样量一致,应根据色谱柱规格相应地放大进样体积。上样量由以下公式确定:
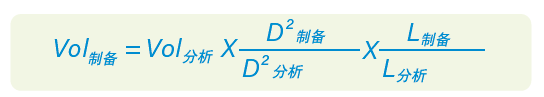
其中,Vol是进样体积(μL),D是色谱柱内径(mm),L是色谱柱柱长(mm) [20]。
类似地,要保持分离效果一致,应根据色谱柱规格放大流速。在色谱柱柱长和粒径相同的条件下,流速的几何放大方法如下:
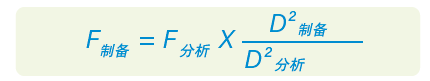
其中,F是流速(mL/min),D是色谱柱内径(mm)20。
延迟体积与柱外体积
当色谱柱柱长相同时,须根据延迟体积调整制备型方法的梯度曲线。要进行上述调整,必须先测定分析型和制备型系统的延迟体积。确定延迟体积的常用方法是向助溶剂中加入能够发出UV信号的化合物或溶剂,并在不连接色谱柱的情况下运行梯度。记录泵处的梯度开始时间与检测器处信号检出时间之间的时间延迟,将时间延迟乘以流速即可确定延迟体积(图32)。
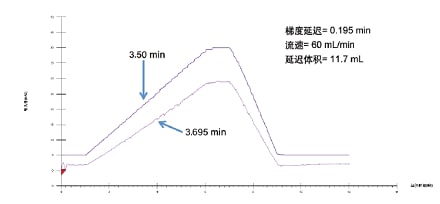
图32. 为确定系统延迟体积生成的数据(在甲醇中加入1%丙酮作为助溶剂)。梯度条件如下:5%保持1 min,在5 min内从5%升至40%,40%保持1 min,在2 min内从40%降至5%,然后5%保持1 min。生成此数据时色谱柱已从系统中移除。
除此之外,还应注意管路、进样阀、定量环、检测器流通池以及分流器等造成的柱外体积影响,其能够导致最终色谱图中出现谱带展宽⁶。在移除色谱柱后执行进样可测定柱外体积和谱带展宽。进样与检测之间的时间差乘以流速等于柱外体积。未连接色谱柱时的峰形可指示所有因柱外体积造成的谱带展宽。图33展示了为测定柱外体积执行的示例进样结果。由于系统采用改性剂流进样的管路设置,我们进样了100%助溶剂,以便在不添加CO2的情况下更准确地测定柱外体积。详细测定方案请通过www.waters.com访问在线应用程序“OBD制备柱计算器”。此色谱柱计算器工具易于使用,可辅助分析级到制备级的所有放大计算。
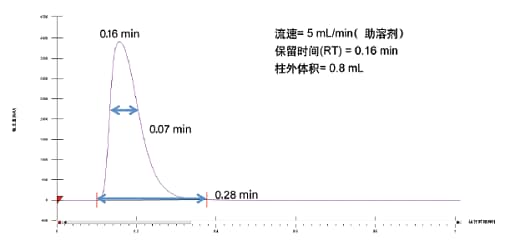
图33. 为确定柱外体积生成的数据。条件如下:以5 mL/min的流速进样100 μL浓度为10 mg/mL的布西丁,采用甲醇方法条件(无CO2),不连接色谱柱。
流动相密度注意事项
尽管简单的LC方法放大规则也适用于SFC,但却不能直接应用,因为这些规则的前提是流动相密度恒定。SFC方法放大更加复杂,主要是由于流动相具有可压缩性,会导致色谱柱内和系统之间出现密度、压力和温度差异。上述因素的变化会影响流动相组成和强度⁹,进而影响保留性和选择性,这进一步增大了使分析型与制备型系统的分离结果保持一致的难度。
虽然无法直接控制密度和温度变化,但我们可以通过调节以下SFC方法参数有效确保不同规模的色谱结果保持一致:
确保系统间平均压力一致(从而使平均密度一致)。
平均压力等于前端压力与背压之和除以2。可通过调整分析型或制备型仪器中自动压力调节器的设置使压力一致,从而使色谱柱内的压力(密度)曲线相似。应注意,平均压力一致不一定能确保密度曲线一致,但其一致性会有所提升。
确保流动相组成(二氧化碳和助溶剂)精确匹配。这基本上是从体积流量到质量流量(分析型系统)或从质量流量到体积流量(制备型系统)的系统间单位换算,目的是得到等效流动相。
对于SFC,助溶剂组分是十分重要的峰保留时间控制参数。因此,准确放大助溶剂组分是方法成功放大的关键。通过匹配CO2及助溶剂的质量流量和组成,以及色谱柱出口的压力和温度,能够可靠地将方法从基于体积流量的仪器放大到基于质量流量的仪器⁹。
方法放大示例
本例中的分析级方法采用UPC2系统开发,参数如下(表8):
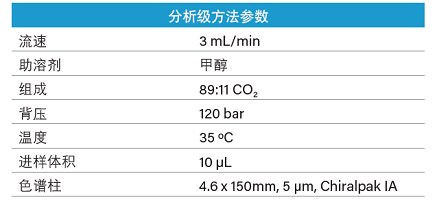
表8. 在制备型SFC系统中进行方法放大所用的优化UPC2分析级方法参数。
为保持色谱柱柱长和粒径一致,制备型系统采用填充5 μm颗粒的21 x 150 mm ChiralpakIA色谱柱。对进样体积进行几何放大,因此内径4.6 mm色谱柱上的10 μL进样相当于内径21 mm色谱柱上的208 μL进样。在本例中,分析型系统的CO2泵基于体积流量实施控制,而制备型系统中的泵基于质量流量实施控制。因此在进行几何放大计算之前,必须先将分析级方法中的CO2流速换算为质量流量。采用如下公式进行换算:
CO2流速= 2.67 mL/min
CO2密度= 0.89 g/mL
CO2流速(质量)= CO2流量 x CO2密度
CO2流速(质量)= 0.89 x 2.67 = 2.38 g/min
由于助溶剂可正常放大(mL到mL)且助溶剂流速为0.33 mL/min,分析级方法的总流速为2.70 g/min(甲醇含量约为12%)。然后对流速和百分比进行几何放大,得到采用21 x150 mm色谱柱时的制备级流速为56 g/min(助溶剂比例为12%)。
流速和流动相组成确定后,还必须通过相同的平均压力使制备型系统的密度曲线与分析型系统相匹配。分析型系统的前端压力为162 bar,背压为120 bar(压降为42 bar),因此可得平均压力为141 bar。由于制备型系统的压降仅为24 bar,因此必须将背压设为130,以匹配分析型系统的平均压力。最后,温度也应相匹配,保持35 °C。采用上述参数,我们成功将分离方法从ACQUITY UPC2放大到Prep SFC系统,如图34所示。所得谱图基本同,但制备型系统中的保留时间稍有延长,可能是进样策略差异所致。分析型系统采用混合流进样,而制备型系统采用的是改性剂流进样。
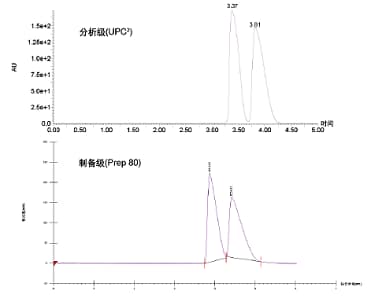
图34. 从UPC2系统到Prep SFC系统的手性分离方法放大示例。
重迭进样
许多应用都采用重迭进样法。重迭进样可缩短进样周期之间的时间,从而将溶剂用量降至尽可能低。重迭进样还可利用所有可用的色谱空间进行连续分离和纯化,能够显著提高通量。重迭进样通常在之前进样的样品仍在色谱柱上(或正从色谱柱中洗脱)时执行。因此,只有采用等度方法时才能使用重迭进样。要成功执行重迭进样,必须确定正确的进样周期(或两次进样的时间间隔)。另外,还需为序列中的最后一次进样确定总运行时间,以确保所有色谱峰组都能洗脱并被收集。图35展示了上述参数的确定方法,并将所得参数应用于一组重迭进样。由于进样周期约为总运行时间的一半,因此在第一个目标峰开始洗脱前执行两次进样。最后一次进样完成后,洗脱并收集到两组色谱峰。在本例中,重迭进样将总处理时间缩短为传统单次进样运行的一半。
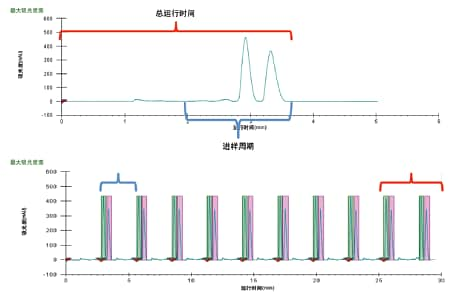
图35. 展示如何根据探索运行结果确定进样周期的色谱图(A),以及将所得结果应用于一组重迭进样所得的色谱图(B)。进样周期以蓝色标识,少于2 min。总运行时间以红色标识,约为4 min。
应用示例
如前所述,SFC有着广泛的选择性范围,适用于多种应用(表9)。
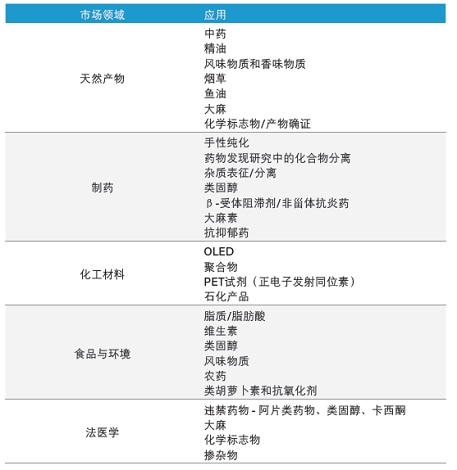
表9:按市场领域分类的制备型SFC应用。
无论在哪个市场领,用于何种纯化用途,Prep SFC都具有以下特性:
通过易于使用的单一平台提供广泛的选择性
可进行各种规模的色谱方法放大
可提高生产效率并节省溶剂
是反相LC的正交技术
可分离和纯化结构相似化合物
本节摘取了两个应用示例,用于展示不同的SFC工作流程及其应用领域。这些应用的完整信息可通过沃特世网站www.waters.com查看。
采用SFC对挥发性风味物质和香味物质进行手性纯化
https://www.waters.com/webassets/cms/library/docs/720005150en.pdf²¹
手性分离是SFC的重要应用领域之一。正如手性药物的不同对映体会表现出不同的药理活性,风味和香味物质的立体化学特性决定了它们的味道、气味品质和强度。由于这些物质具有挥发性,纯化的难度非常大。SFC为手性风味和香味物质的纯化提供了一种快速、低温且高回收率的方法。
在本例中,我们采用重迭进样的手性Prep SFC方法分别从薰衣草和茶树精油中纯化芳樟醇对映体和松油烯-4-醇对映体。图36所示为4.6 x 250 mm AD-H色谱柱到半制备型10 x250 mm AD-H色谱柱的分析级方法放大。茶树精油分离结果显示其含有两种松油烯-4-醇对映体,而薰衣草精油中只含芳樟醇对映体中的一种。Prep SFC方法参数见表10。
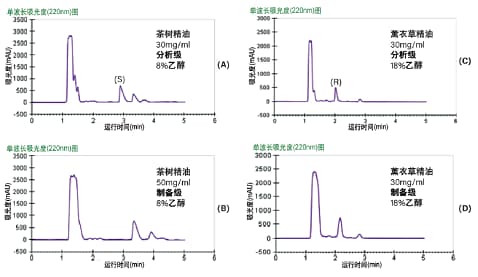
图36. 在等度条件下采用经过优化的分析型和制备型分离方法分析茶树精油(A和B)和薰衣草精油(C和D)所得的结果。
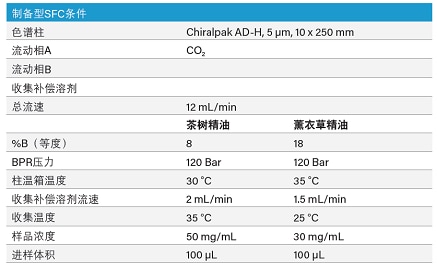
表10. 用于从茶树精油和薰衣草精油中纯化松油烯-4-醇和芳樟醇的制备型SFC方法条件。
该方法为等度方法,因此采用了重迭进样,以期大幅提高收集效率。根据色谱分析结果,茶树精油的进样周期约为3 min,薰衣草精油的进样周期约为2 min。采用重迭进样得到的手性纯化结果见图37,其中50 mg茶树精油在40 min内完成了纯化,30 mg薰衣草精油在30 min内完成了纯化。
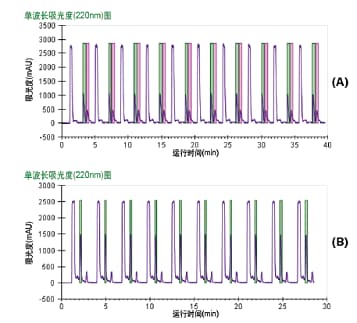
图37. 茶树精油(A)和薰衣草精油(B)重迭进样和收集得到的色谱图。
馏分分析结果见图38,其中全部三种馏分的纯度均大于92%。我们使用松油烯-4-醇和芳樟醇的外消旋标准品进行了回收率研究,结果表明回收率可达70%~80%,鉴于现有文献报道的回收率都极低,该结果已非常出色。
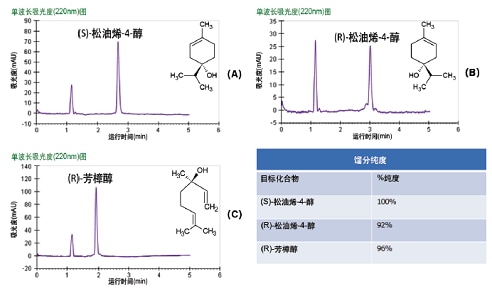
图38. 茶树精油和薰衣草精油的馏分分析结果。
应用基于MS的馏分触发机制对药物化合物进行非手性纯化
https://www.waters.com/webassets/cms/library/docs/720005064en.pdf22
采用UV引导的纯化时,由于许多化合物的吸收波长相同,检测器将无法区分色谱峰。质谱引导的纯化基于化合物质量数收集馏分,该参数可有效区分目标化合物与任何杂质,因此更具针对性。活性药物成分合成后,终产物中可能存在中间体杂质。
Imatinib是一种用于治疗多种癌症的酪氨酸激酶抑制剂。在本例中,我们纯化了Imatinib合成的反应中间体和终产物。第一步,使用三种手性色谱柱,采用添加或不添加氨水添加剂的甲醇对混合物分离方法进行筛选。筛选结果如图39所示。BEH 2-EP色谱柱以及添加了氨水添加剂的甲醇被选为理想方法参数用于优化和放大。
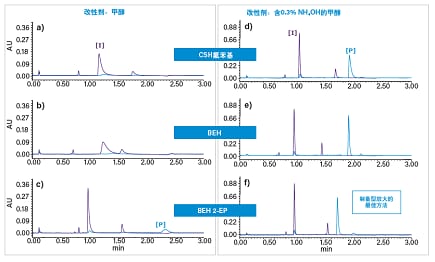
图39. 初始色谱柱筛选实验,分别使用甲醇(左)和含有0.3% NH4OH的甲醇(右)作为助溶剂。色谱柱为粒径1.7 μm的2.1 x 50 mm ACQUITY UPC2色谱柱。筛选梯度为2 min内从4%升至40%,流速1.5 mL/min。温度为40 °C,背压设定为1800 psi。
为了优化放大后的分离效果,我们分别为中间体和产物建立了梯度聚焦。根据筛选梯度的斜率和各目标峰的保留时间,计算出洗脱时的助溶剂浓度。中间体洗脱时的助溶剂比例为14%,产物洗脱时的助溶剂比例为为29%。以上述比例为中心建立2-min梯度聚焦,起始点为助溶剂比例-5%,结束点为助溶剂比例+5%(图40)。
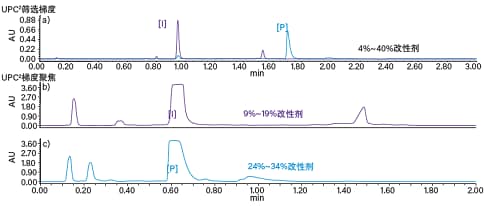
图40. 中间体(I)和产物(P)的色谱图(以含有0.3% NH4OH的甲醇为助溶剂,采用BEH 2-EP色谱柱)。梯度条件:(a) 2 min内助溶剂从4%升至40%;(b) 2 min内助溶剂从9%升至19%,用于纯化中间体(I);(c) 2 min内助溶剂从24%升至34%,用于纯化产物(P)。
放大方法时采用相同填料的色谱柱,同时保持柱长与粒径的比值(L/dp)不变。将方法从1.7 μm颗粒的3 x 50 mm分析型色谱柱(L/dp = 29.4)放大到5 μm颗粒的19 x 150 mm制备型色谱柱(L/dp = 30)。放大后的色谱方法(梯度聚焦)和质谱引导的馏分收集结果见图41。图42所示为Imatinib产物和中间体馏分的分析结果。
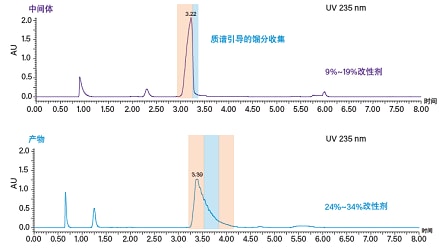
图41. 放大后的制备型色谱方法(采用质谱引导的收集)得到的中间体和(上图)和产物(下图)谱图。分离采用含0.3% NH4OH的甲醇作为助溶剂,梯度用时5.1 min。分别采用助溶剂比例为9%~19%和24%~34%的梯度聚焦执行中间体和产物的纯化。
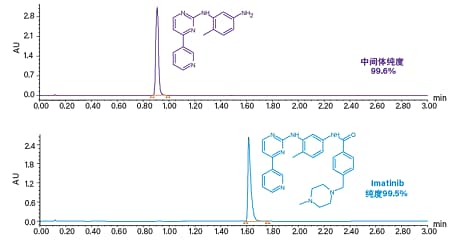
图42. 中间体(上图)和Imatinib产物(下图)的馏分分析结果。采用初始分析级筛选参数,助溶剂梯度为2 min内从4%升至40%。
