ALTERNATIVE METHOD DEVELOPMENT
If an isocratic separation or a focused gradient does not adequately resolve the compound of interest, the separation can be redeveloped through changes in the solvent, pH, stationary phase and/or temperature. These types of modifications can have a dramatic impact on the separation, therefore scouting gradients followed by method optimization need to be performed after any of these changes are made.
SOLVENT
HPLC mobile phase is comprised of three components; weak solvent, strong solvent, and modifier. Solvents must be of high purity, compatible with the detector, nonreactive with the sample, and have low viscosity to keep system backpressure low.
In reversed-phase chromatography, the weak solvent is almost always water, while acetonitrile and/or methanol are commonly used as the strong solvent due to low viscosity, high strength, and UV favorability in the low wavelength range. Acetonitrile and methanol also provide the best peak shape, are easy to evaporate after isolation, and tend to be nonreactive with most samples.
As with any solvent used for chromatographic separation, it is important to remain above the reported solvent UV cutoff value during sample analysis. Below this value, changes in the solvent composition are recorded by the detector as baseline drift or other chromatographic disturbances.
|
Mobile phase
|
UV cut-off
|
|
Water
|
200 nm
|
|
Acetonitrile
|
190 nm
|
|
Methanol
|
205 nm
|
|
Ethanol
|
210 nm
|
|
Propanol
|
210 nm
|
|
Isopropanol
|
205 nm
|
Table 2: Common chromatographic solvents and UV cut-off.
Separation and peak order can be different depending upon the strong solvent used for the separation. The solvent that will provide the highest resolution for the compound of interest is difficult to predict, therefore, the choice for strong solvent is often determined by trial and error. Optionally, strong solvents can be mixed (e.g. 50:50 acetonitrile / methanol) to achieve a desired elution order or resolution.
pH
Mobile phase pH is a very important variable in the control of retention for reversed phase separations. Compounds often contain one or more acidic or basic functional groups therefore most reversed-phases mobile phases require pH control.
When an acid is more than 2 pH units above or below its pKa, it will be >99% ionized or unionized, respectively. In contrast, bases are ionized below their pKa and non-ionized above their pKa. The unionized form will be less polar (more hydrophobic), and thus more strongly retained in a reversed-phase system. As a result, acids are better retained at low pH, whereas bases are better retained at high pH.⁴
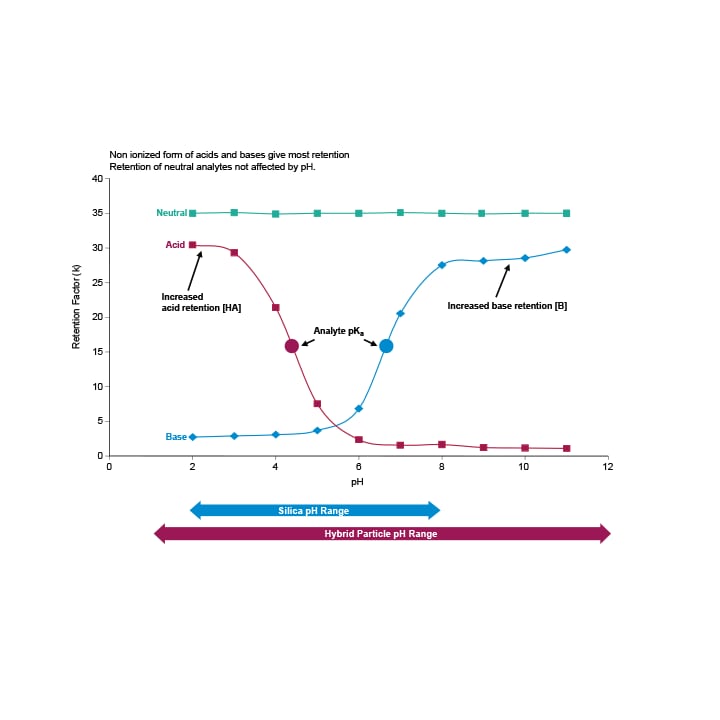
Figure 10: Effect of mobile phase pH on analyte retention. For the most robust separations, choose a mobile- phase pH that corresponds to the plateau regions of the retention map. Non-ionized form of acid and bases give the most retention while retention of neutral analytes are not affected by pH.
If the mobile phase pH is near the pKa of the compound of interest, small changes in pH can make large changes in retention, which can directly impact the robustness of the separation. pH of the mobile phase is controlled by the addition of a buffer. The buffer maintains the pH when a small amount of acid or base is added. It is most effective when used within ±1 pH unit of its pKa, but may provide adequate buffering ±2 pH units from the pKa.
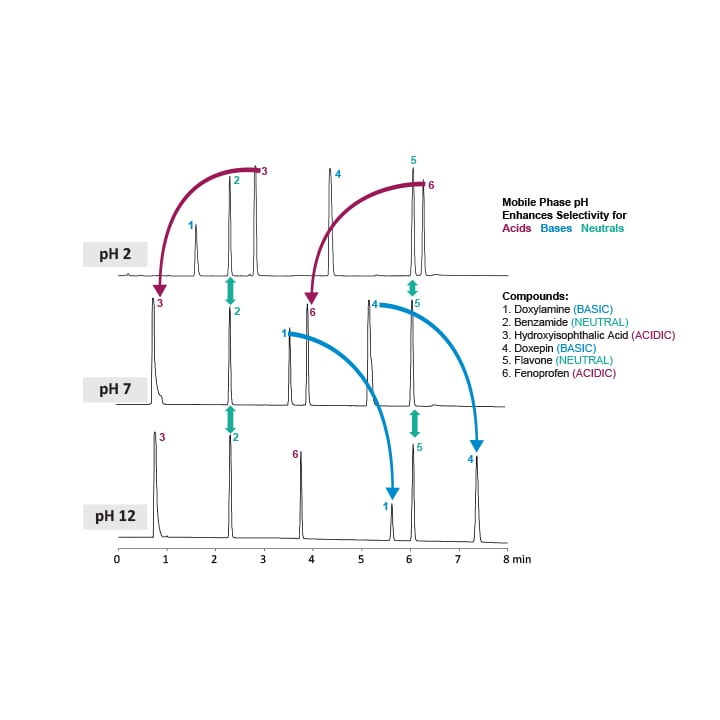
Figure 11: Compound selectivity vs. pH. Elution order dramatically changes based upon the ionization of acidic and basic compounds, while neutral compounds are unaffected.
When selecting a working pH, the stability of the column should also be considered.
Silica-based columns are best operated between pH 2 and pH 8. A bonded phase is susceptible to hydrolysis at low pH and at high pH the silica backbone becomes increasingly soluble. At pHs above 8, a non-silica based particle, or a particle with a ligand specifically engineered for high pH stability, is required. pH limits and general handling recommendations can be found in the column package insert, or on the website of the column manufacturer.

Figure 12: Example of the Waters Charged Surface Hybrid (CSH) silica based engineered ligands and the suggested pH range.
When a chromatographic method is intended to be employed for purification purposes, the buffer additives used to adjust the pH must be adequately volatile to facilitate removal from the purified isolate. Additionally, when using volatile additives, care must be taken to avoid contamination or precipitation in the MS source. Common additives such as formic acid, acetic acid, and ammonium acetate work best at a concentration of 0.05–0.1% when dissolved in the mobile phase. Phosphate, the most popular buffer additive for liquid chromatographic separations, is not recommended for purification or MS applications.
Buffer concentrations between 5–10 mM are recommended. When using buffers, it is important to filter the buffer after preparation, and flush the pump lines when not in use to prevent in-line precipitation, and replace the solutions regularly to prevent build-up of biological growth.
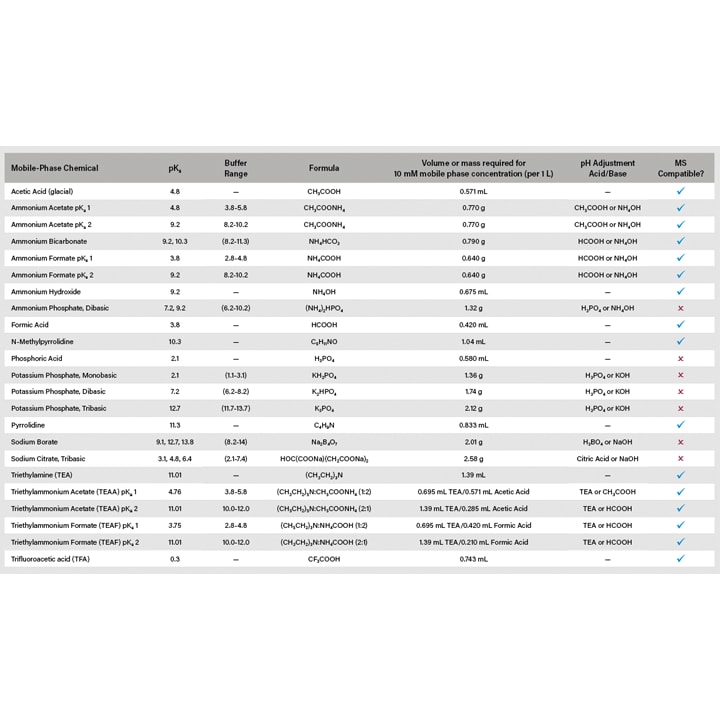
Figure 13: Mobile phase buffer selection guide with MS compatibility.
STATIONARY PHASE
The column stationary phase has a significant impact on the separation. Stationary phases for reversed phase range from C18 to C8 or can contain engineered ligands to promote selectivity and separation power. Many laboratories have a limited column inventory therefore replacing the stationary phase is often a final resort when solvent and pH do not provide adequate resolution. The Waters Reversed Phase Column Selectivity Chart (www.waters.com) provides a selectivity comparison between different column stationary phase manufacturers. This tool is very useful in early method development to compare column selectivity.
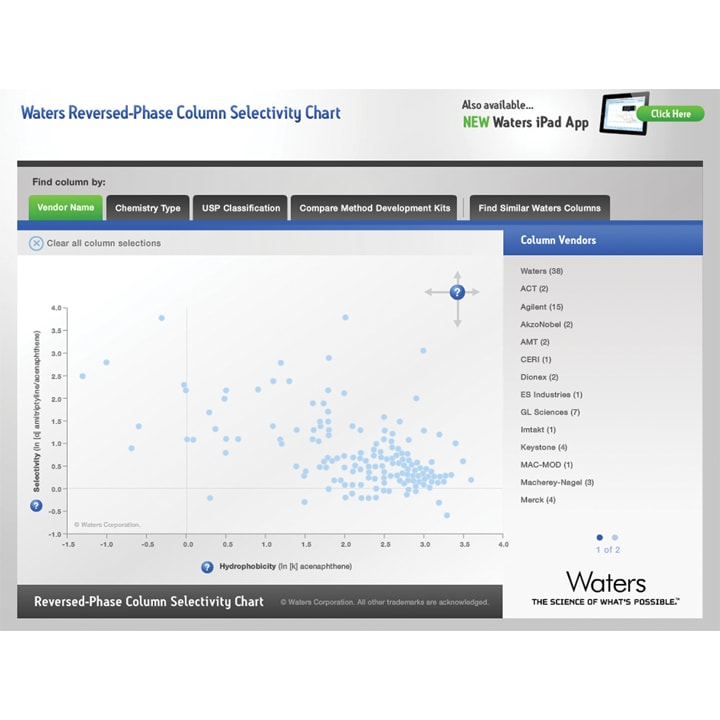
Figure 14: The Waters Reversed-Phase Selectivity Chart available in the Web Toolbox at www.waters.com/selectivitychart.
LENGTH
Column length is a factor in separation performance. Columns can be purchased in a range of lengths including 25 mm, 50 mm, 100 mm, 150 mm, and 250 mm.
Shorter columns have lower plate counts, but are good for fast separations, while longer columns have higher plate counts and result in longer retention times. With longer length, backpressure, run time, solvent consumption, and cost proportionally increase, therefore the shortest column that provides adequate resolution is the most ideal option.
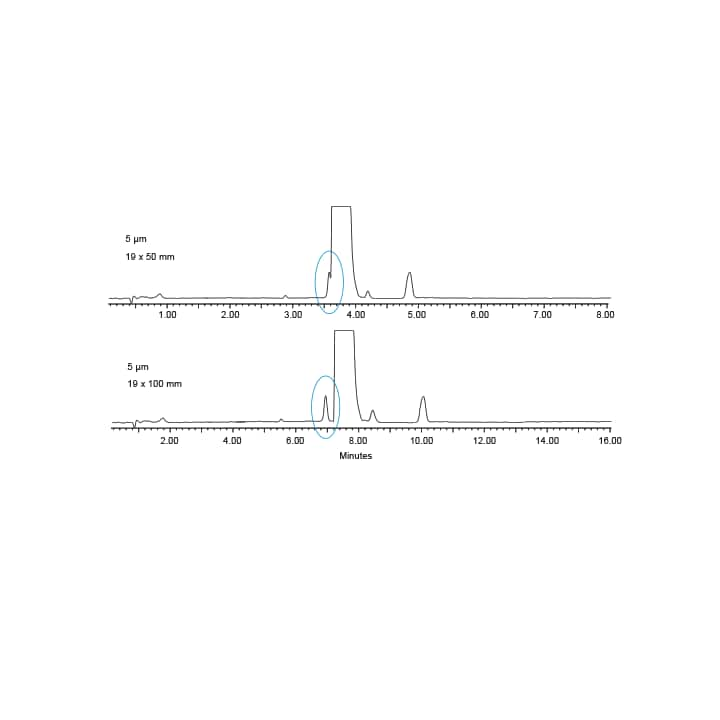
Figure 15: Comparison of resolution for column length. The 100 mm column shows increased resolution of the main peak and the proceeding impurity. The total run time increased with an increase in column length.³
Particle Size
The column's ability to resist the dispersion or spread of a sample band as it passes through the packed be is called "efficiency." Smaller particle sizes increase efficiency because peak dispersion is lower within the particles. High efficiency results in narrow peak widths and better separation power.
Prep column particles sizes typically range from 5–10µm, while ultra-high pressure analytical separations use 1.7–3.5µm particle sizes. Although very small particle sizes provide greater efficiency, the trade-off is an increase in column cost and system back pressure. Large scale prep columns, which run at high flow rates with crude sample mixtures, are often not offered in very small particle sizes for these reasons.
Temperature
Since different temperatures can impact selectivity, column heating is a convenient tool for optimizing the separation of a particular sample. Mobile phase viscosity and total system pressure are reduced with elevated temperature, which results in less strain on the system, fittings, and column, to ultimately increase the ruggedness of the operation. Temperature can also decrease retention times and change the selectivity of the separation. It is difficult to predict whether temperature changes will increase or decrease peak resolution, therefore the utility is specific to each particular separation.
While temperature control in small scale chromatography is routine, temperature is infrequently employed as a parameter for manipulating chromatography intended for prep scale purification. First, large diameter columns can be difficult to heat uniformly from the outside. Second, high flow rate separations used for large scale tend to remain at the temperature of the incoming solvent. Electric blankets and column ovens, while satisfactory for small scale separations, cannot heat a large column uniformly across the diameter of the column. As a result, temperature gradients form across the diameter of the column, adversely affecting the chromatography.
If temperature control is necessary to maintain sample solubility, temperature gradients can be overcome by placing the prep column in a heated water bath with a length of tubing plumbed at the head of the column. The length of tubing acts as a preheater to equilibrate the incoming solvent at the desired temperature.⁵ Chromatography intended for future scale-up, is best developed under ambient conditions and temperature control employed as a last resort.
