ISOLATION OF THE PURIFIED PRODUCT
After a separation method has been developed and scaled up to the desired scale, the final step is to isolate or collect the compound of interest. Fractions can be collected manually by the operator as peak elution is monitored, or fraction collection can be automated based on detector signal or retention time.
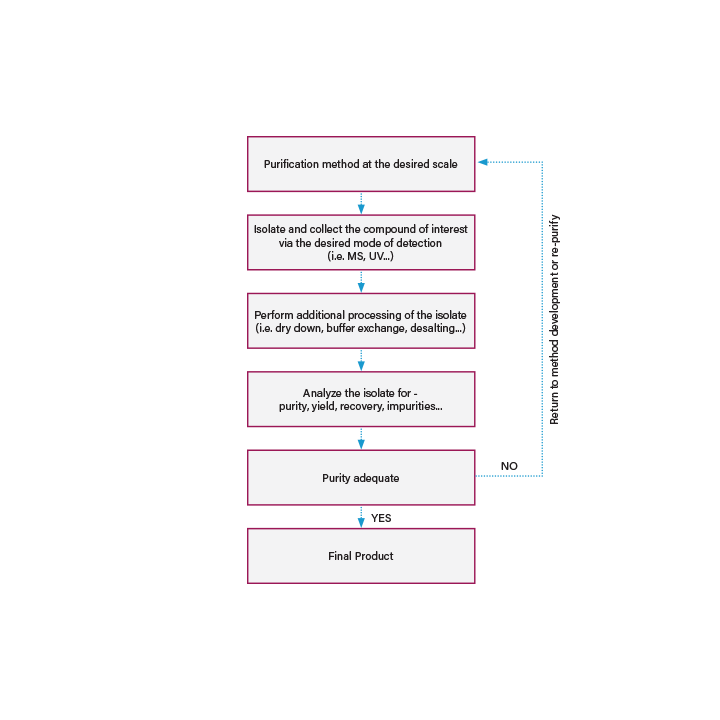
Figure 41: Workflow for isolation of the purified product.
FRACTION DELAY TIME
Fraction collection occurs when the detector sees a peak that meets user defined collection triggering criteria. When the peak is present in the detector, it has not yet traveled to the fraction collector. If a fraction collector is triggered too soon, the peak would be missed by the fraction collector. A system specific, fraction delay time must be determined beforehand in order to time fraction collection correctly. One method of calibration involves injecting a concentrated dye and comparing the time it takes to be seen by the detector, to the time it takes to reach the fraction collector. Fraction delay time calibration is usually a one-time process and once entered into the software it can be re-calculated for any flow rate. Fraction delay time must be re-calibrated when changes are made to the flow path from the detector to the fraction collector, such as when a flow splitter is added.
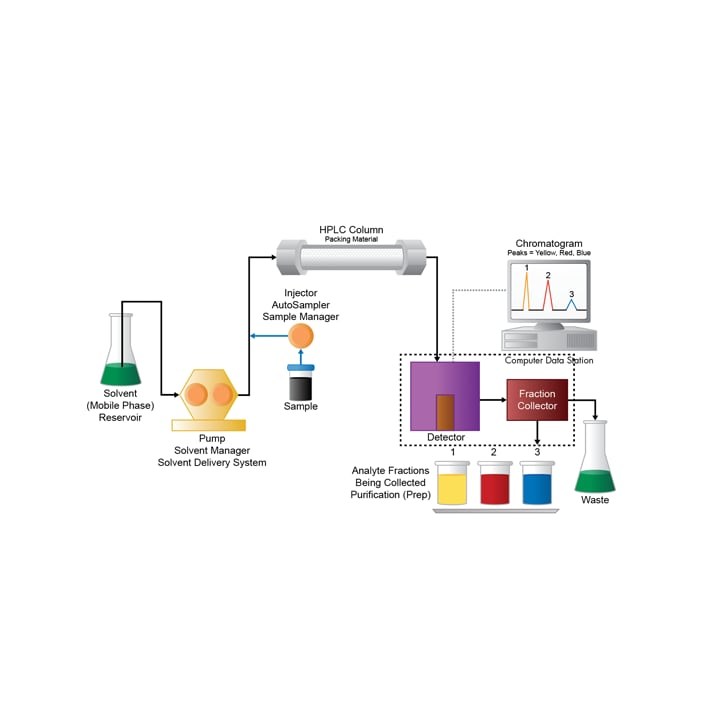
Figure 42: System flow segment circled that contributes to fraction delay time.
MANUAL FRACTION COLLECTION
Manual collection is quite straight forward and easy. The user triggers the switching of the diverter valve on the fraction collector while the elution of the compound is visually monitored through an online based instrument software real-time signal plot. Manual fraction collection provides the highest flexibility because only the desired parts of the purification run are collected, but throughput tends to be low due to the lack of automation.
PEAK BASED FRACTION COLLECTION
Peak based triggering is an automated way of collecting fractions based on a detector signal. Parameters used to decide whether a peak in the chromatogram should trigger the fraction collector are usually peak height, threshold, and/or slope.
The most common approach for peak-based fraction collection is based on height or threshold. As soon as a signal reaches a predefined detector height, fraction collection is triggered. When the signal falls below a predefined height, collection is stopped.
Fraction collection by slope is often a more challenging method of collection, but it can be used to collect peaks that are not baseline separated. When collecting by slope, fraction collection is automatically triggered by the software at mathematical inflection points detected along first derivative of the chromatogram.
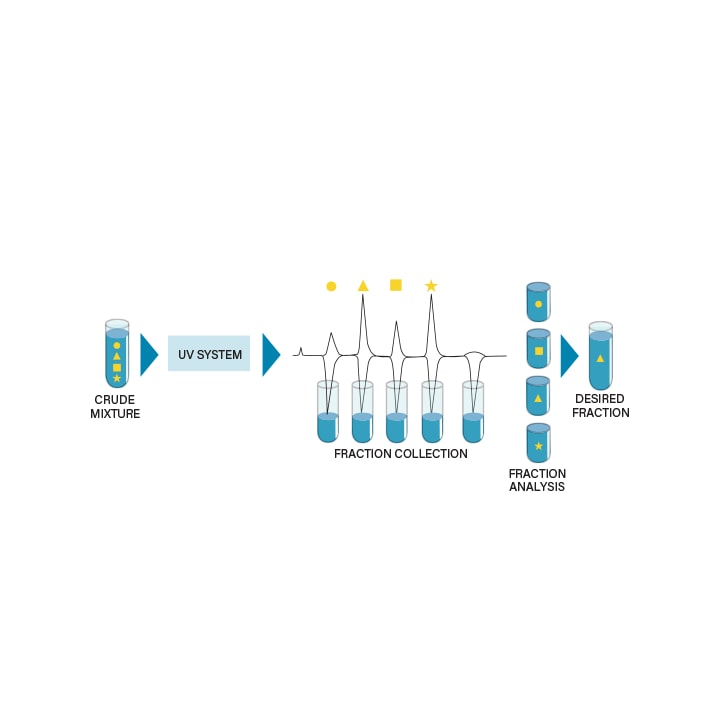
Figure 43: Peak based fraction collection.
FRACTION COLLECTION BY MASS DETECTION
In mass based fraction collection, only the compounds with the user defined desired mass are collected. As a result, collection by mass can be more efficient than peak- based collection. The requirements for mass-based fraction collection are summarized as follows:
■ The molecular mass of the compound must be known.
■ The compound must ionize for mass detection.
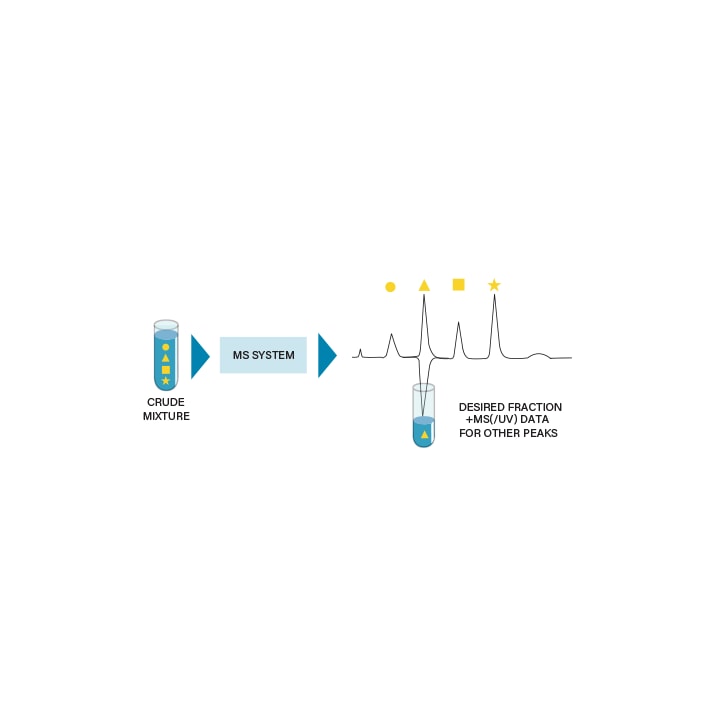
Figure 44: Mass based fraction collection.
FRACTION ANALYSIS
Once the fractions have been collected, they can be analyzed directly in the collection vessel or the solvent can be removed to yield the purified product. A rotary evaporator (rotavap) can be used to bring the fraction to dryness. With this tool, the solvent is evaporated from the sample and the aqueous portion is freeze dried. If inorganic buffers have been used or if the aqueous portion is particularly large, the fraction can be passed through a reversed phase SPE cartridge to trap the target compounds (i.e. known as desalting). The trapped compounds are then eluted with a small amount of organic solvent, which can be evaporated off more easily.¹

Figure 45: Rotary evaporator set-up.
An estimate of recovery can be determined by fraction analysis using various routine analytical techniques such as; UV, IR, MS, NMR, X-Ray, assay, and/or structure elucidation. When a standard is available, it is easy to make a direct comparison of the isolate with literature data, however if the target compound is unknown, a broad, comprehensive systematic approach involving a variety of physical, chemical, and spectroscopic techniques is required to establish a purity and stability profile.
Equation 14: % Recovery
When analyzing the isolate, if the purity, recovery or activity is not what is expected, evaluate the isolate for the following situations:
■ When testing directly from the collection vessel, concentration gradients can form. As a result, the purity result is dependent upon where the sample is drawn from in the collection vessel. Samples should be well mixed before analysis to achieve a representative result.
■ If the sample is most soluble in a strong solvent such as DMSO, but not in mobile phase, the collected fraction will crystallize as it is stored in the collection vessel over time. If the liquid phase is drawn for purity and concentration analysis, the results will only be representative of the soluble portion.
■ The active compound can also be lost due to decomposition between the time of collection, the purity test, and the activity test. It can also decompose during the dry-down process. Therefore, activity testing is almost always performed before and after dry-down.
■ The active compound has been retained on the column.
■ The active compound is unstable in the conditions used in the isolation process.
■ The extract solution may not have been prepared in a solvent that is compatible with the mobile phase, so precipitation may have occurred.
■ Most of the active compound is spread across a wide range of fractions causing undetectable amounts of the compound present in the fractions.
■ The activity of the extract could be due to the presence of other compounds in the crude sample and not active individually.¹
■ The isolate may not be as pure as anticipated if the isolate was collected at a UV wavelength where close eluting impurities did not absorb.
If the isolate is not within the purity, throughput or recovery requirements of the application, a re-purification of the isolate can be performed either using the same separation method, or a new overall separation method can be developed with different column selectivity. It is up to the chromatographer to determine the time and effort they wish to invest for the purification to meet the desired purity, throughput and recovery objectives.
