Case Study: Investigating Unexpected Results of a Global Cross-Laboratory Study of a USP Organic Impurities Method on an Arc HPLC System
Abstract
In a global economy many laboratories need to transfer methods around the world. Transferring a method requires a coordinated approach and strategy to control the variables associated with the method, including instrumentation availability, sample prep, mobile phase preparation, etc. However, regardless of the strategy, it can be difficult to control variability in laboratories around the world, especially considering geographical, environmental, and linguistic differences. In the event of any outlier or unexpected results, a good understanding of the factors that can impact the method are important for investigation and resolution. In this application note, a global inter-laboratory method transfer of a USP organic impurities method was conducted across eight sites around the world. Key information for the control strategy and method performance was obtained from the original site and implemented into the study. A further in-depth analysis of the control strategy can be found in the application note “Successful Global Cross Lab Method Transfer of a USP Organic Impurities Method to an Arc HPLC Using a Risk-based Approach”.1 With the control strategy in place, the system suitability requirements were met routinely for all the laboratories; however, the understanding of the method and system variables provided guidance into investigation and cause of unexpected results.
Benefits
- The Arc HPLC System provides easily replicated, quality data performance from system to system
- Employing the XBridge C8 Column reduced the column-to-column variability when transferring the USP Quetiapine Impurities method from system-to-system
- The traceability function in the Empower 3 Software provides a record that each lab involved in the study has conformed to the instructions outlined appropriately
Introduction
In a global economy many laboratories need to transfer methods around the world. Transferring a method requires a coordinated approach to control the variables associated with the method, including instrumentation availability, sample prep, mobile phase preparation, etc. However, regardless of the strategy, it can be difficult to control variability in laboratories around the world, especially considering geographical, environmental, and linguistic differences. In the event of any outlier or unexpected results, a good understanding of the factors that can impact the method are important for investigation and resolution.
In this application note, a global interlaboratory method transfer of a USP organic impurities method was conducted across eight sites around the world. Key information for the control strategy and method performance was obtained from the original site and implemented into the study. With the control strategy in place, the system suitability requirements were met routinely for all the laboratories; however, the understanding of the method and system variables provided guidance into investigation and cause of unexpected results. These unexpected results will be looked at in depth in this application note.
Experimental
The method was based on the USP monograph for Quetiapine Fumarate Impurities,2 with no adjustments.
Sample Description
The method requires both a system suitability reference standard (RS) and a quetiapine fumarate RS. The system suitability solution was prepared from the USP quetiapine system suitability RS (USP p/n: 1592715) and consists of a mixture of quetiapine, quetiapine desethoxy (1–5%), related compound G, and related compound B. The system suitability solution was prepared at 1 mg/mL in Diluent (86:14 Mobile phase A/Mobile phase B).
The standard solution was prepared utilizing the USP quetiapine fumarate RS and was prepared at a concentration of 0.001 mg/mL in Diluent.
The drug substance was obtained from Hangzhou Think Chemical Co., Ltd. and past the date of expiration. The sample was prepared at 1.0 mg/mL in Mobile phase A.
Method Conditions
|
Column: |
XBridge C8 3.5 µm, 4.6 x 150 mm (Waters p/n: 186003055) |
|
|
Column temp.: |
45 °C |
|
|
Sample temp.: |
10 °C |
|
|
Injection volume: |
20 µL |
|
|
Detection: |
250 nm |
|
|
Data rate: |
10 Hz |
|
|
Flow rate: |
1.5 mL/min |
|
|
Run time: |
70 minutes |
|
|
Buffer: |
3.1 g/L Ammonium acetate in water. Add 2 mL of 25% Ammonium hydroxide to each 1 L of solution. pH = NLT 9.2 |
|
|
Mobile phase A: |
25:75 ACN:Buffer |
|
|
Mobile phase B: |
Acetonitrile |
|
|
Needle wash: |
50:50 Water:acetonitrile |
|
|
Purge solvent: |
50:50 Water:acetonitrile |
|
|
Seal wash: |
90:10 Water:acetonitrile |
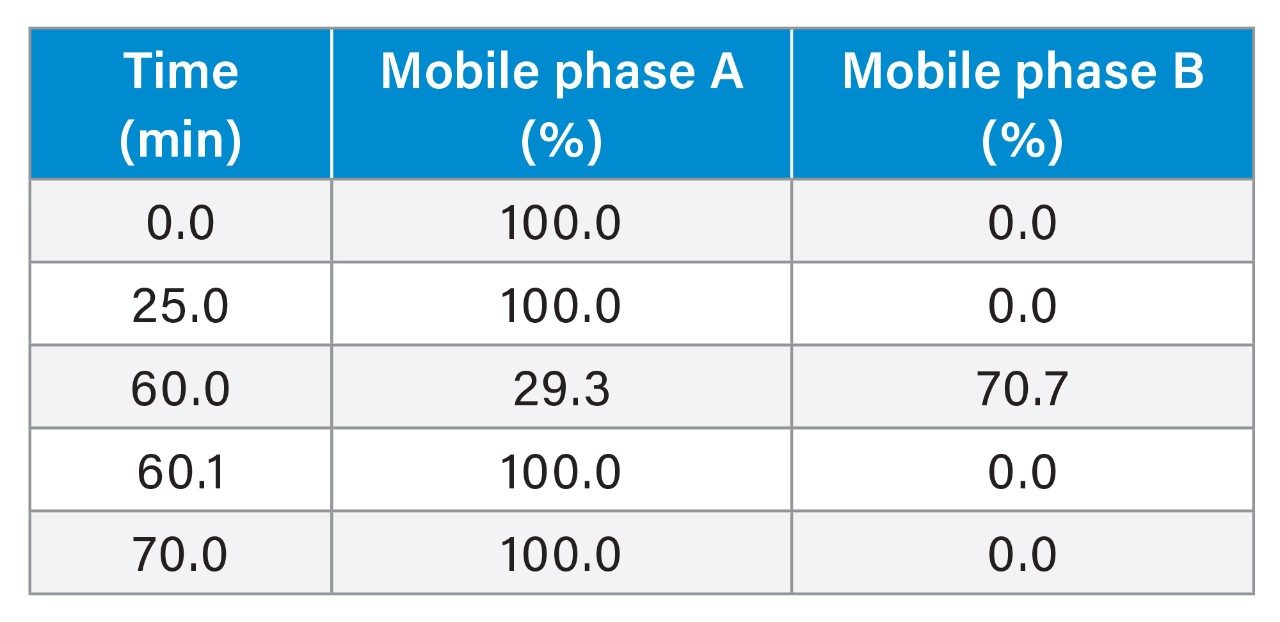
Gradient
Data Management
Empower 3 Chromatography Data Software
System Configuration
|
System: |
Arc HPLC System (QSM-R, FTN-R, and CHC) |
|
|
Detection: |
2998 (PDA) or 2489 (TUV) |
|
|
Configuration: |
Passive preheater (preferred) |
|
|
Flow cell: |
Analytical |
Results and Discussion
The Study
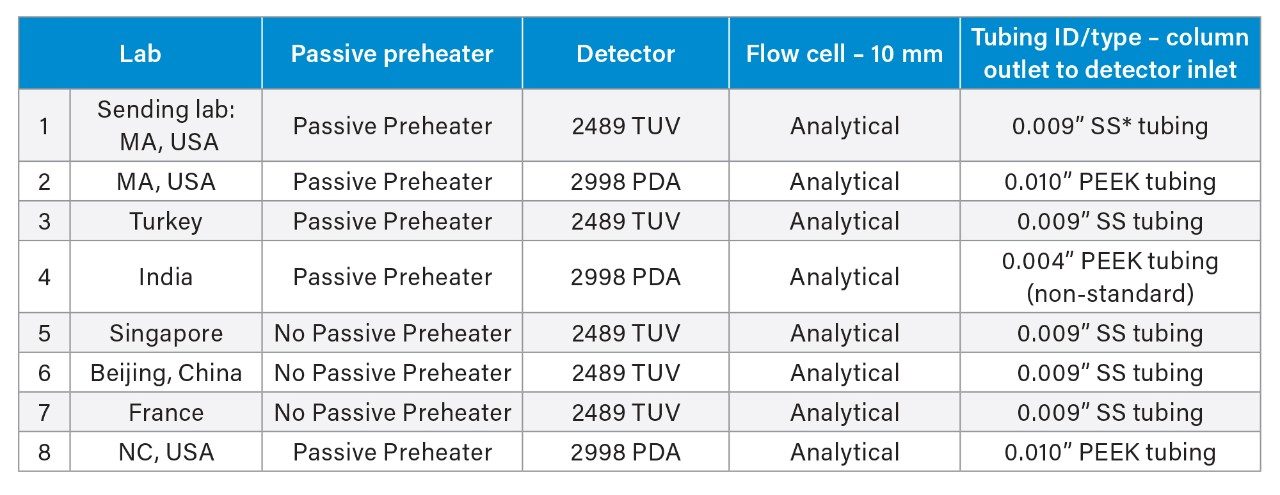
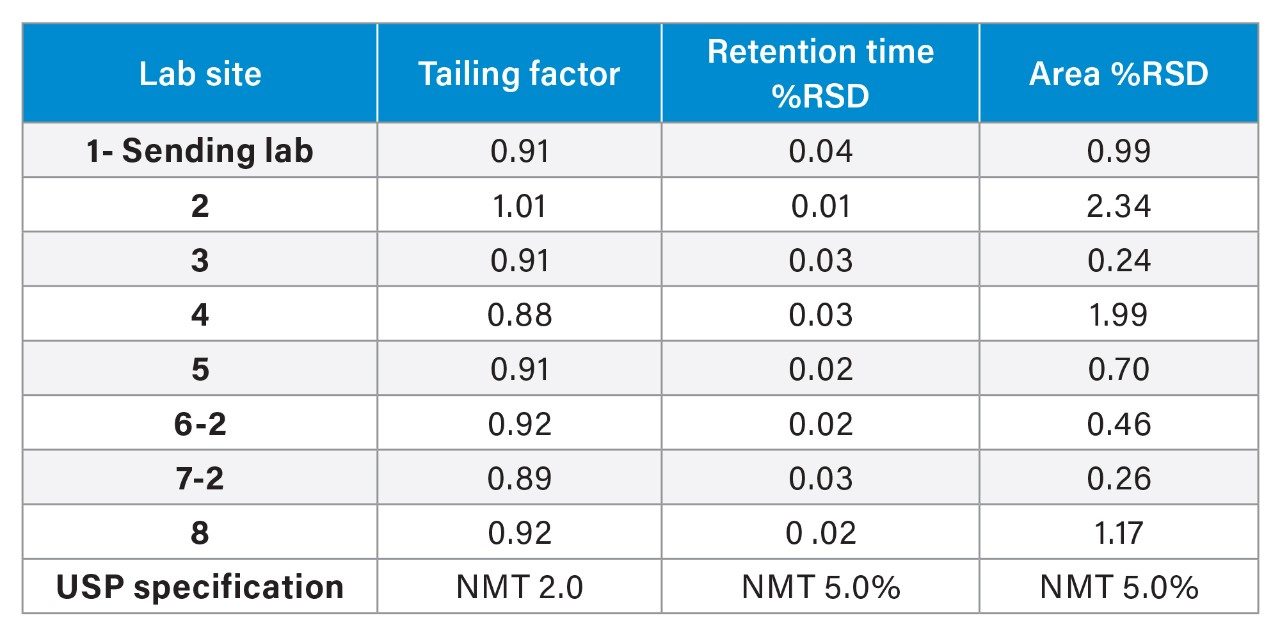
Method transfer of the USP Quetiapine Fumarate Impurities method2 was conducted across sites around the world. The laboratories were located in Milford, MA, USA (sending laboratory) and 7 receiving labs – Milford, MA second lab, Turkey, India, Singapore, China, France, and North Carolina, USA (Table 1). The study included verifying the system suitability requirements and quantitative analysis of a drug substance. All analyses were performed on the Arc HPLC System with either a Tunable Wavelength (TUV) 2489 Detector or a photodiode array (PDA) 2998 Detector. Each analysis was assessed using the system suitability requirements, as described in the monograph, as well as analyzing the drug substance and comparing the impurity analysis. The system suitability criteria were based on the resolution of two critical pairs in the system suitability solution, the tailing, retention time %RSDs, and area %RSDs from the standard solution.
For the study, control strategies were setup to ensure the reproducibility of the method around the world. For example, a single chemical kit was sent out to all the laboratories involved, which included the standards, the column, and the sample formulation. (Note: Not all the columns were from the same lot.) A Standard Operating Procedure protocol was written and distributed to the many labs. To maintain the original USP method, preparation of the mobile phase, including filtration of the mobile phase and the use of new or recently opened containers of chemicals involved in the method, for example the ammonium hydroxide and the ammonium acetate, were carefully described. For more information on these control strategies and how they were implemented, see the application note “Successful Global Cross Lab Method Transfer of a USP Organic Impurities Method to an Arc HPLC Using a Risk-based Approach”.2
The quetiapine impurities method was baselined in the sending laboratory and then transferred to the remaining laboratories. Each lab processed the data to ensure that system suitability was met, however, all the quantitative data was processed at the sending laboratory site using a single processing method. When all the data was reviewed, there were some inconsistencies and unanticipated results that needed to be examined further.
Case Study 1: Retention time differences
Problem
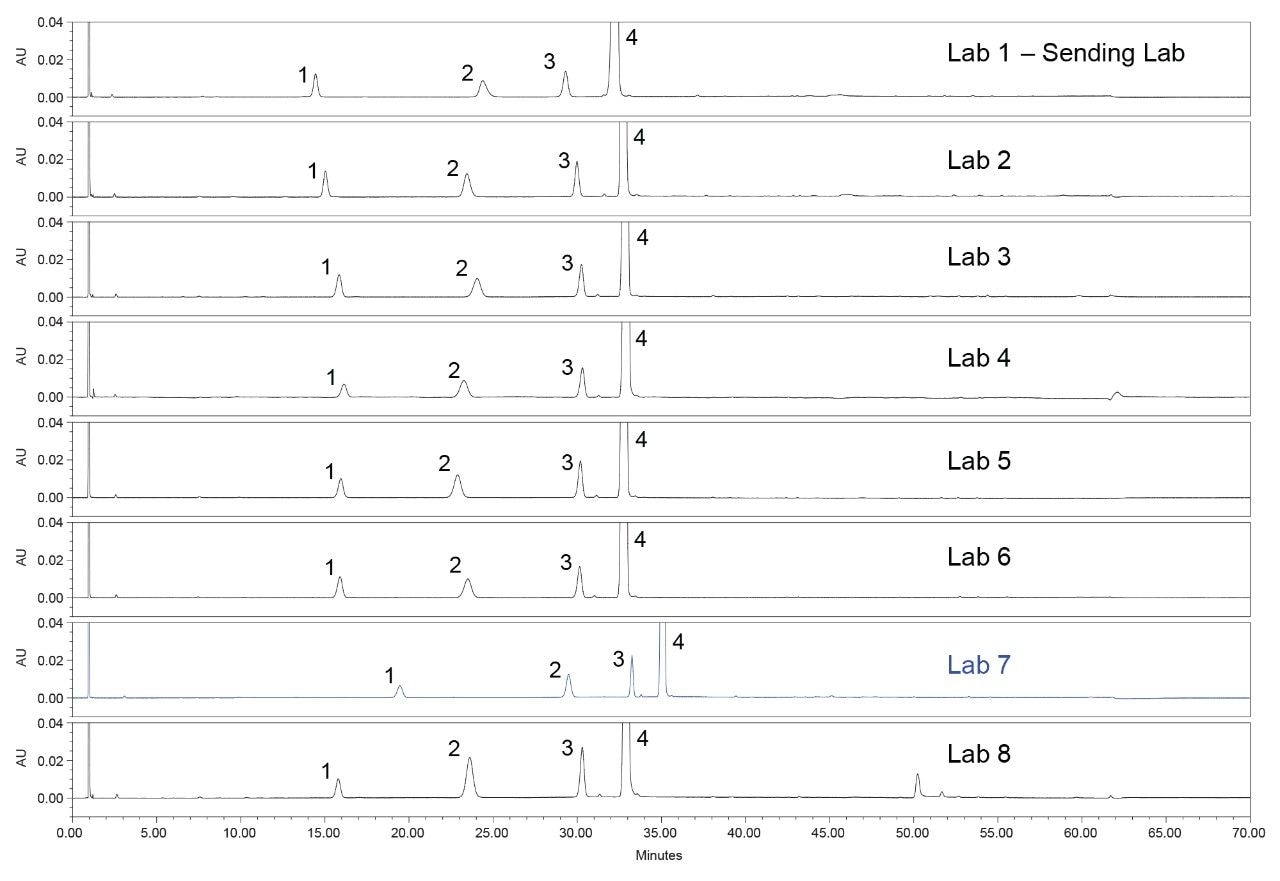
The system suitability solution measures the resolution of two critical pairs, quetiapine (API) and quetiapine desethoxy and the resolution of Related Compound G and Related Compound B. All eight laboratories from the study met requirements for the two critical pairs and the chromatograms can be seen in Figure 1. When analyzing the chromatograms, the results from all the laboratories had reproducible retention times, within 1%. However, one lab, Lab 7, produced chromatography with a significant retention time shift as compared to all the other labs. Although Lab 7 passed the system suitability requirements, an investigation was conducted to determine the possible cause of the retention time shift.
Investigation
- Instrument
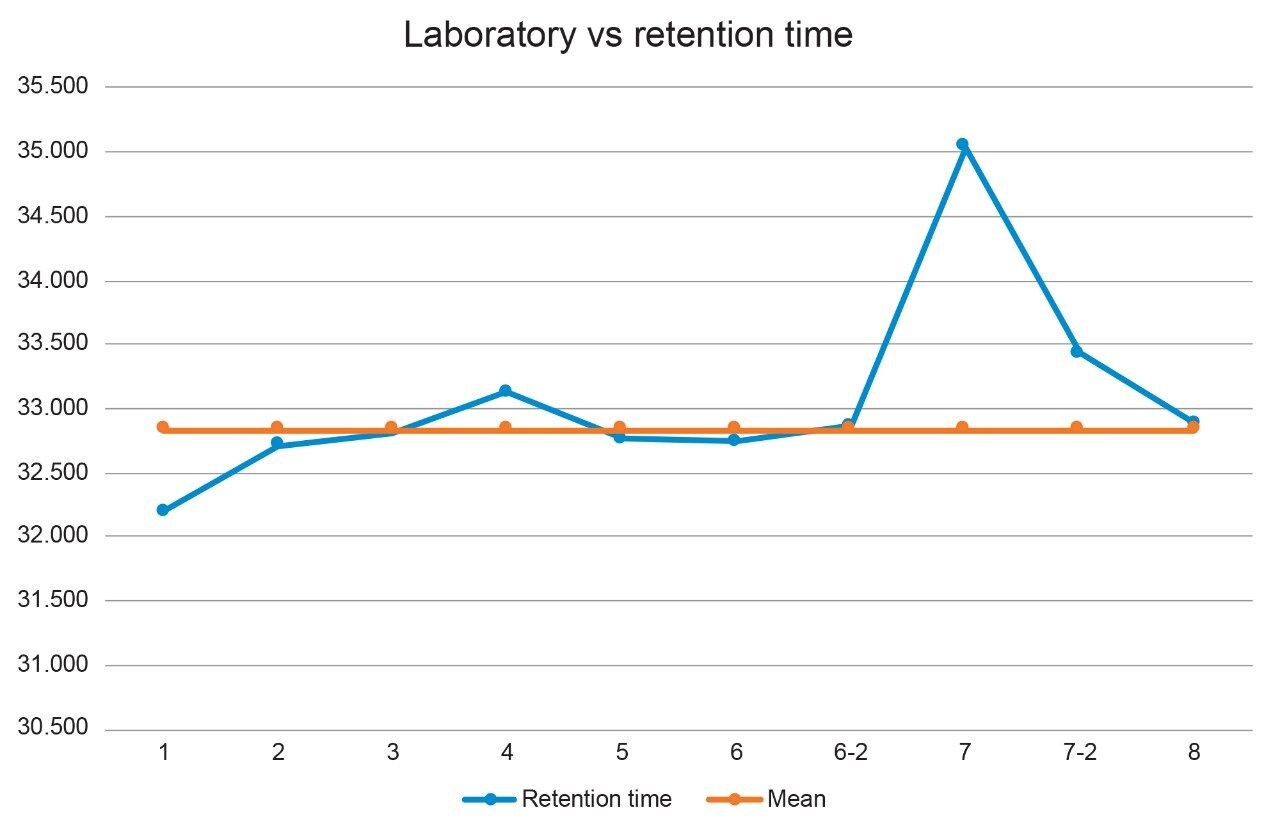
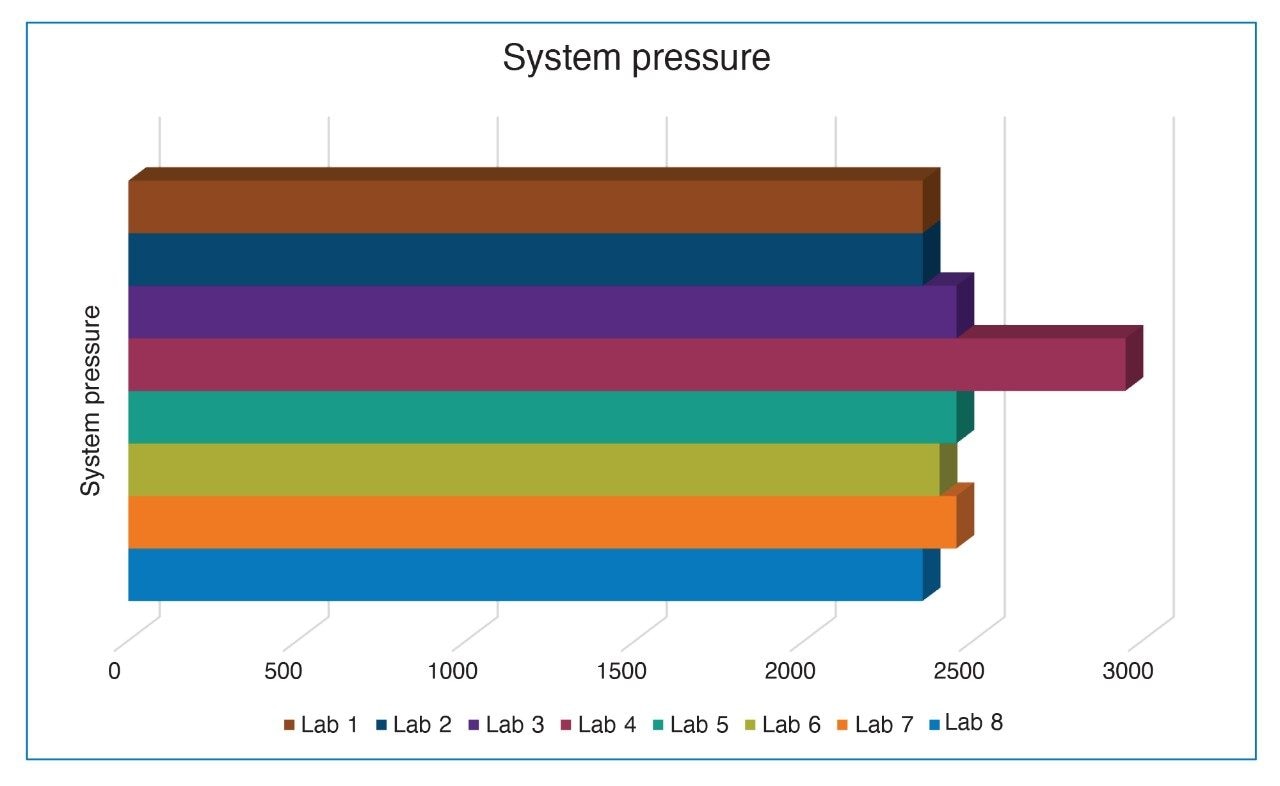
When the retention times for the quetiapine peak for all laboratories was plotted (Figure 2), it was observed that Lab 7 had a retention time shift of over two minutes. The first step taken to determine the cause of the retention time shift was to look at the system pressure trace from each of the systems (Figure 3). If the pressure of the system from Lab 7 was greater than the other laboratories, the increased pressure might have affected the retention time. The system pressure for Lab 7 was comparable to the other labs, therefore it was not the system pressure causing the retention time shift.
However, when investigating the system pressure, Lab 4’s Arc HPLC System was found to have a substantially higher backpressure than all other systems and labs, with a system pressure approximately 20% higher. Investigation of the systems found a correlation in the system configurations. While all the other labs used the standard configuration tubing of 0.009” or 0.010” from the column outlet to the detector, Lab 4’s system was configured with non-standard tubing with an ID of 0.004” (see Table 1), between the column outlet and the detector. The impact of the lower ID tubing is both decreased resolution and higher back pressure. Therefore, there is no correlation between the pressure of the system and the retention time of the peaks. However, all system suitability requirements were comparable as seen throughout the application note.
- Procedure/Mobile Phase Prep
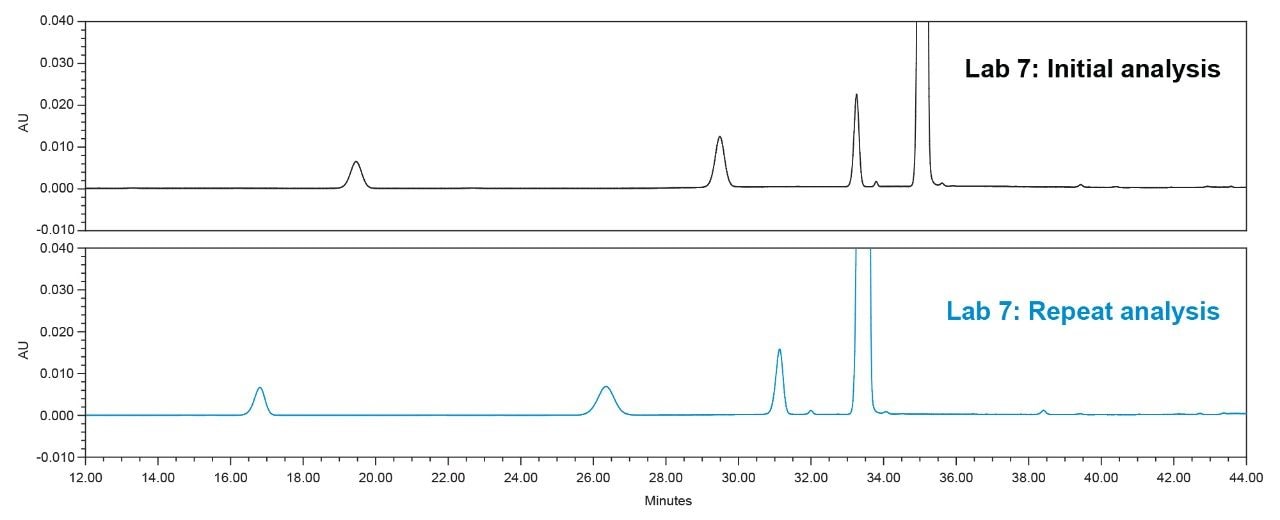
By ruling out the system pressure as a possible source of the retention time shift, more information was needed to determine the root cause; therefore Lab 7 repeated the analysis (Figure 4). The second analysis showed the retention of the peaks were much closer to the other laboratories, as can be seen in Figure 2 above for Laboratory 7-2, indicating that the mobile phase was a potential cause of the retention time shift. Also, it was realized that Lab 7 did not have a calibrated pH meter; therefore, the exact pH of the buffer solution was unknown. For the first analysis, Lab 7 added 4 mL’s of ammonium hydroxide, as instructed by the SOP, but for the second analysis the lab added 5 mL’s of the ammonium hydroxide to the buffer solution because it was believed that the pH was not at the desired value of pH 9.2. Therefore, the pH of the buffer solution may be one cause of the retention time shift. Another potential cause could be the organic to buffer ratio for Mobile phase A of the mobile phase based upon a previous study that is not shown in this paper. As expected for reversed phase methods, any relative increase in acetonitrile content will shift peaks to earlier elution times, whereas a relative decrease in acetonitrile will shift peaks to a later retention time.
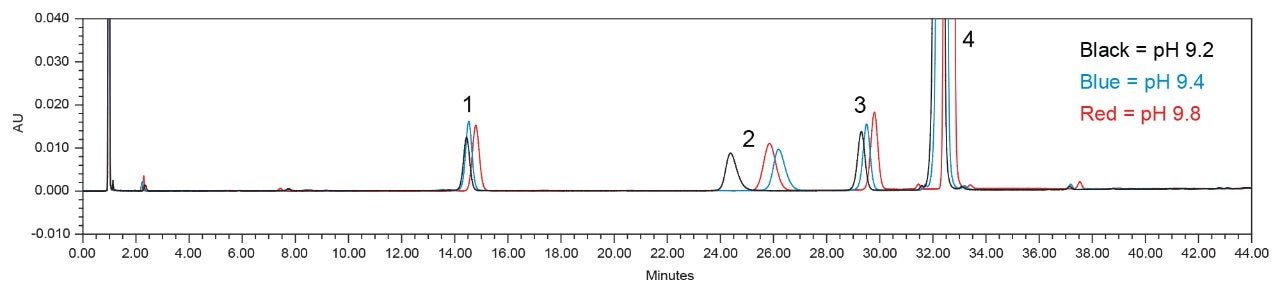
To determine if the pH was affecting the retention time, the sending laboratory conducted a test to determine the effect of the pH on the retention time of the peaks. When regularly running the method in the sending laboratory, the pH of the buffer solution is 9.2 after addition of the required 4 mL of ammonium hydroxide. The sending laboratory added 5 mL’s of ammonium hydroxide to the buffer solution, which gave a measured pH of 9.4. An additional buffer solution was prepared at pH 9.8, which required addition of approximately four more milliliters of ammonium hydroxide. After each of the different pH buffer solutions were prepared, Mobile phase A was prepared as described in the monograph and then analyzed to assess the impact of pH 9.2, pH 9.4, and pH 9.8 on retention time. The resulting chromatograms for the system suitability solution run with the different pH values can be seen in Figure 5. As can be seen in Figure 5, the pH does affect the retention time of the peaks but not as significantly as the retention time shift initially seen from Lab 7. Based upon this information and previous studies looking at the organic composition of Mobile phase A, the hypothesis is that the organic to buffer ratio was most likely the cause of the retention time shift.
- Solution
To reduce variability from mobile phase preparation, it is important to measure out the mobile phase solutions accurately and precisely in defined apparatus for the quetiapine impurities method. Although it is impossible to completely remove the variability between analysts, it is possible to reduce some of the variability by specifying the exact size and the grade of the apparatus in which the mobile phase is prepared. Minimizing the variability of preparation from solution to solution provides better reproducibility.
Case Study 2: Area %RSD Differences
Problem
The standard solution is used to measure the system suitability requirements of tailing factor, retention time %RSDs and area %RSDs. As described in the application note titled “Successful Global Cross Lab Method Transfer of a USP Organic Impurities Method to an Arc HPLC Using a Risk-based Approach”,1 the system suitability results (Table 2) were well within the system suitability requirements. However, in reviewing the results, three data points appeared to be outliers, specifically a higher percent area RSD was observed for Lab 2, Lab 4, and Lab 8. Even though these values met the requirements, an investigation was performed to better understand the root cause.
Investigation
With the system configuration information obtained from the laboratories, it was observed that the three laboratories with the higher %RSDs used a PDA Detector. The chromatograms from the three PDA detectors were compared to the TUV Detector results and it was observed that there were more perturbations in the PDA data than the TUV data (Figure 6). The reduced noise in the baseline on the TUV is more uniform and not unexpected given that TUV detectors have higher sensitivity and a wider linear range than PDAs. The greater baseline noise seen with the PDA results in more area variability for the standard solution, which is at a concentration of 1 μg/mL.
To investigate the hypothesis, the quetiapine impurities method was tested on a single Arc HPLC System with the two individual detectors. First, the analysis was performed on an Arc HPLC with a TUV (2489) Detector, and then the detector was swapped out for a PDA (2998). When comparing the results (Figure 7), the PDA Detector showed greater noise than the TUV Detector on the single Arc HPLC System. Also, the results (Table 3) indicated a significant difference in area %RSD while the other results for tailing factor and retention time %RSD were comparable. The data indicates the baseline noise differences are due to the detector and not the sample, mobile phase, or system variability. While the TUV performed better for this analysis, both detectors met the system suitability requirements specifications.
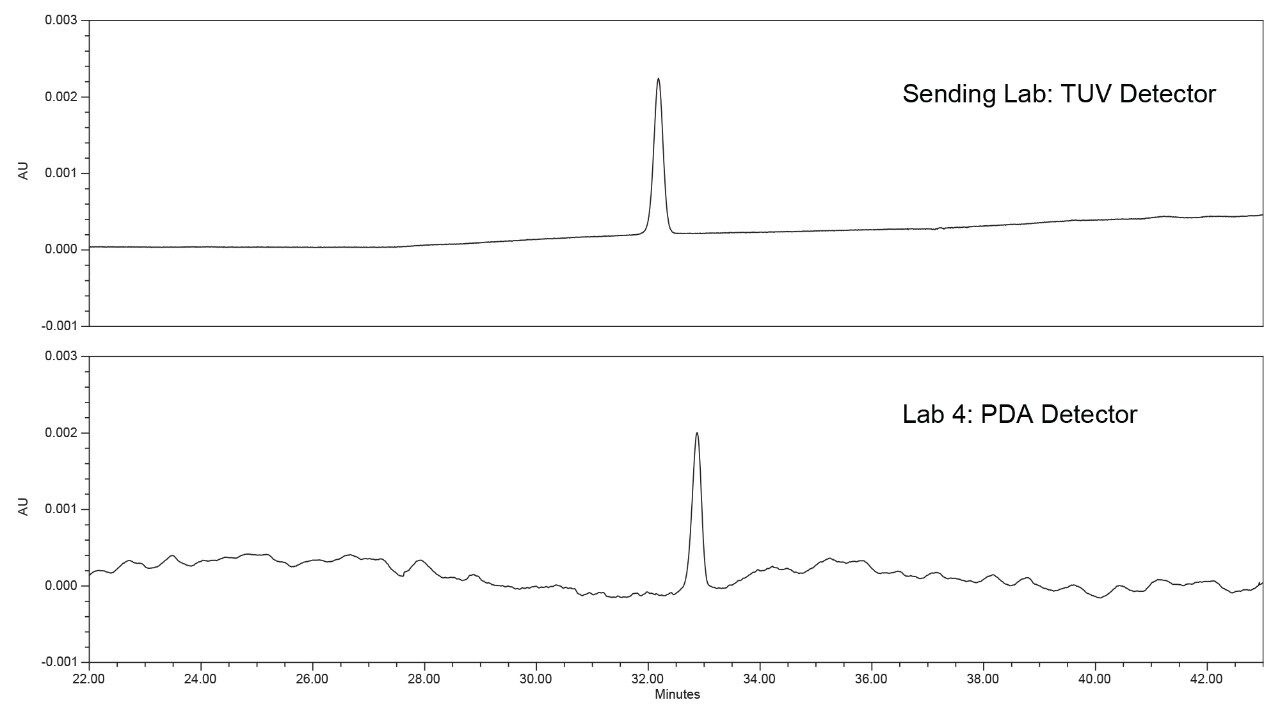
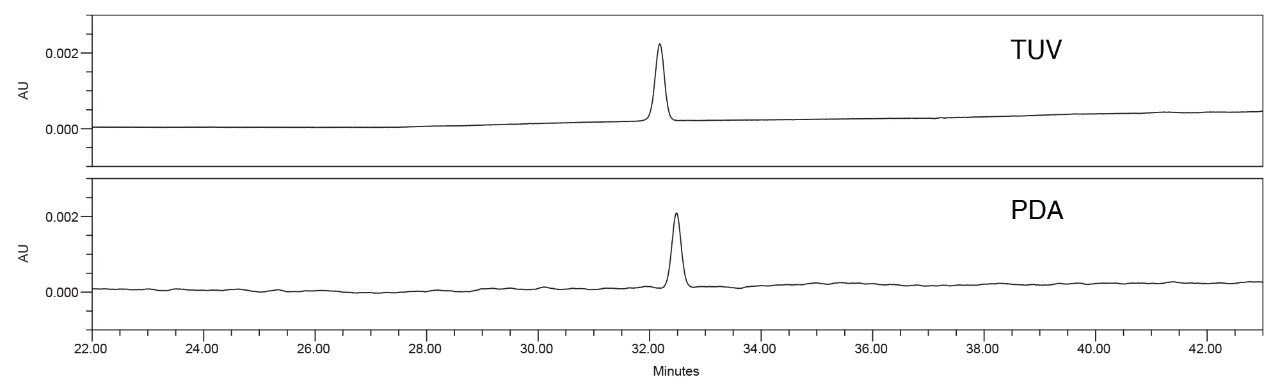
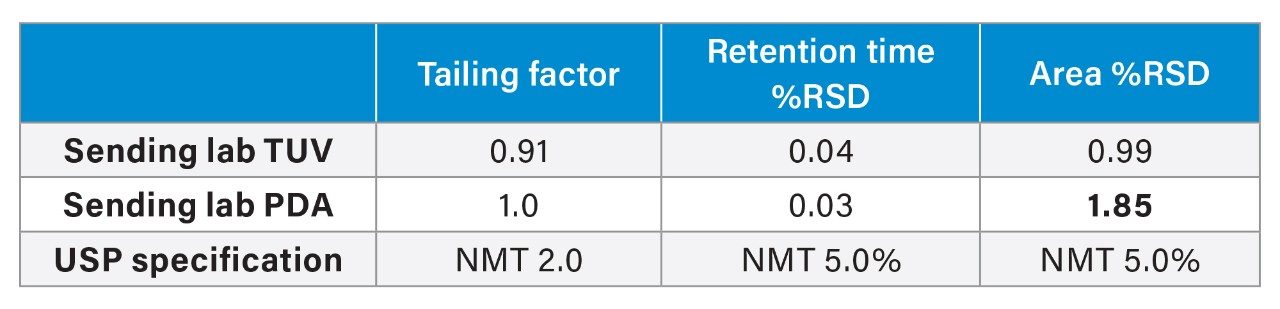
Solution
For future studies a possible solution or approach would be to test samples at lower concentrations on both a TUV and a PDA, ensuring both detectors meet sensitivity or precision requirements. In this example, in a normal troubleshooting experiment, the greater area %RSD’s would normally have been attributed to the injector or other component, but by investigation of the system configuration first a trend was observed. The example shows how investigation of the entire system is warranted regardless of the chromatographic results. The results also demonstrate the need to assess a method on both PDA and TUV if a specific detector will not be required. This ensures, as in this case, both detectors can meet the system suitability requirements of the method.
Case Study 3: Difference in Quantified Results
Problem
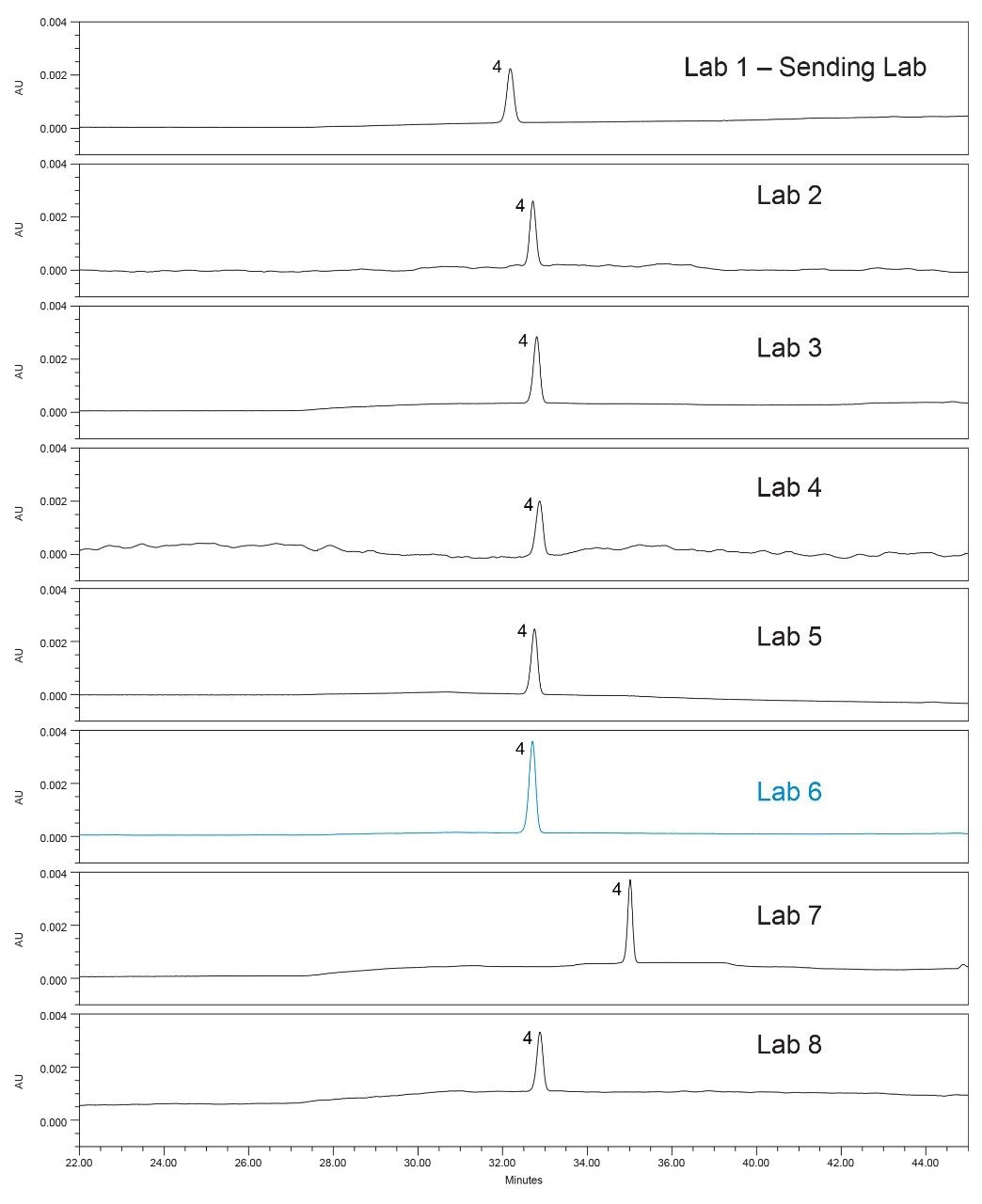
The sample solution or drug substance was analyzed for the quantitative results of the impurities quetiapine desethoxy and of an unknown impurity. All the labs, except one, had comparable quantitative results (Table 4). In this example, upon review of the data, the % of two impurities (quetiapine desethoxy and an unknown impurity) were much lower values for Lab 6. Due to these results, a closer look at the data was taken to determine the cause of the lower impurity values.
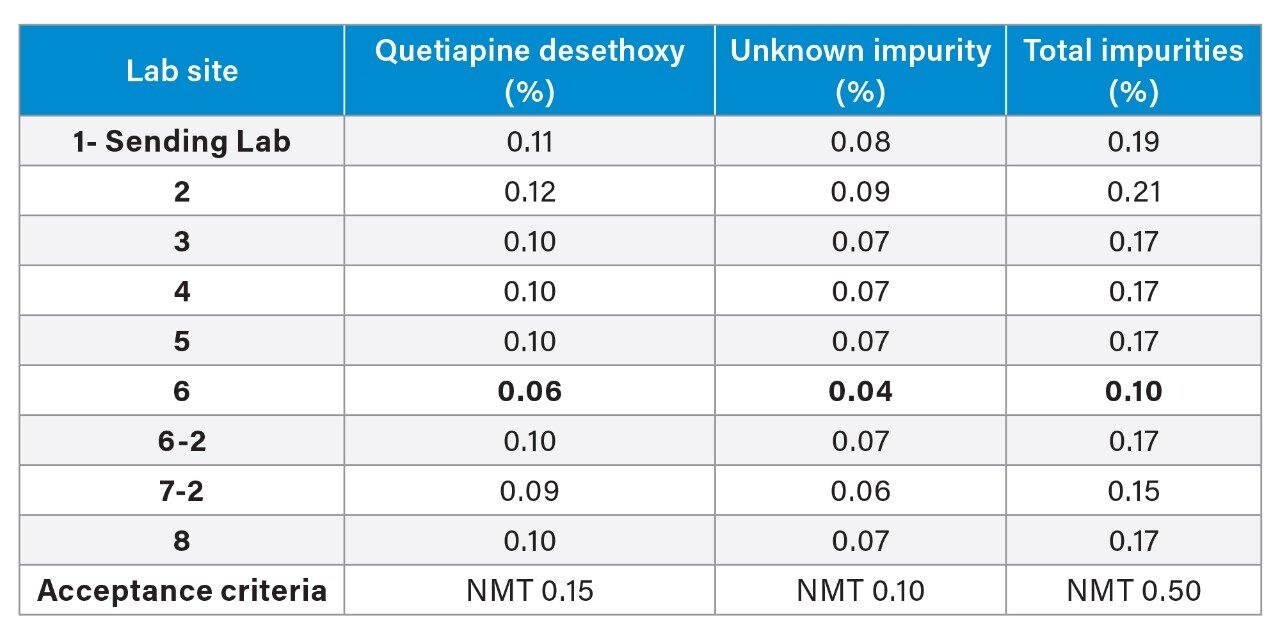
Investigation
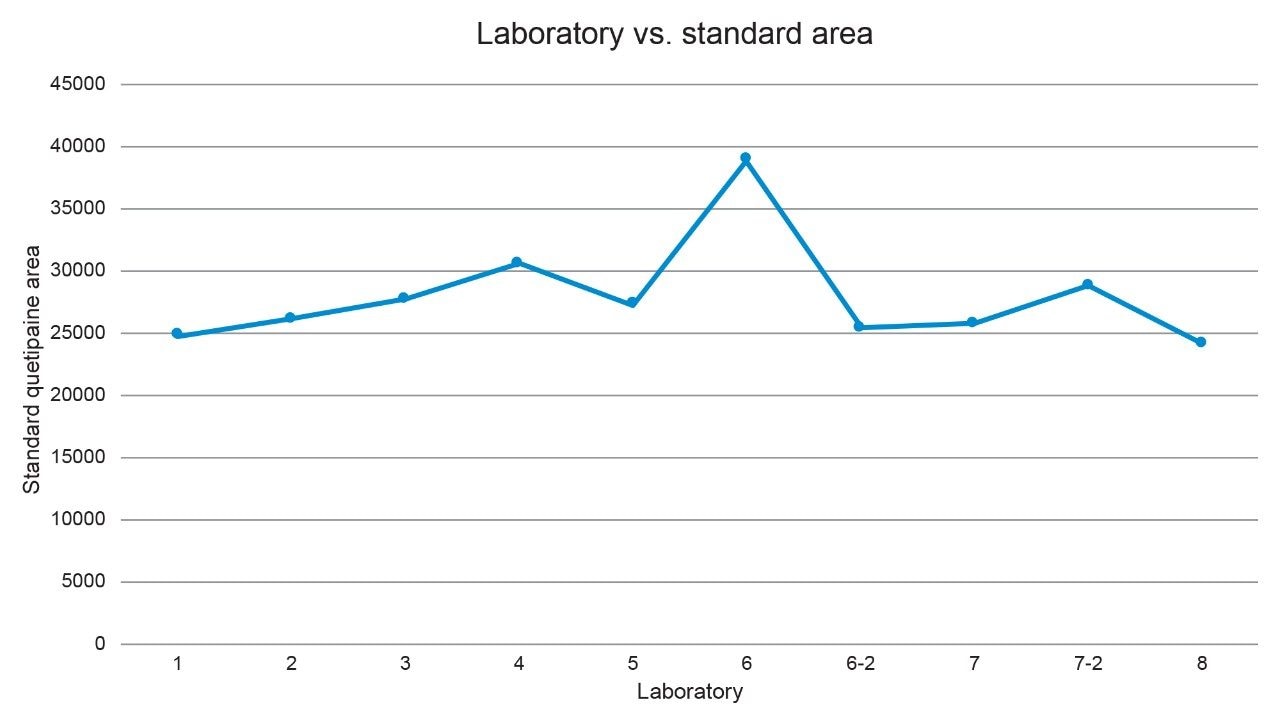
The first step taken was to compare the standard chromatograms from all the laboratories (Figure 8) and compare the standard peak areas from each lab (Figure 9). The standard chromatogram was investigated because the sample impurity amount is calculated relative to the standard solution as a percent of the standard area. It was observed that the standard peak area from Lab 6 was greater than all the other labs. To determine the cause for the greater area, the standard preparation was investigated. First, the concentration of the standard was confirmed to be within 0.2% as described in the SOP, based on review of documentation between the sending and receiving lab. With no clear root cause, the lab was asked to repeat the analysis and re-prepare the standard solution and the sample solution. The standard solution for the second analysis had a comparable peak area to the other labs, as can be seen in Figure 9 for Lab 6-2. In addition, when using the re-prepared standard and sample solutions data, the calculated percent impurity was equivalent to the values obtained from the other labs (Table 4). Therefore, the percent impurities calculated across all systems were now comparable. The standard sample preparation was identified as the likely cause.
Solution
The solution to this error is to change the SOP standard stock solution preparation to minimize the variability that was seen in the study. In the original SOP, weights in the single mg’s were used, however, these low weights increase the likelihood that the sample preparation variability might impact the quantitative results. By increasing the weight used for the stock solution, the variability from analyst-to-analyst and laboratory-to-laboratory could be reduced.
Conclusion
The USP impurities method for quetiapine fumarate was successfully transferred globally on the Arc HPLC System and met all system suitability requirements. While variation in results was observed across the sites, the investigations showed that the cause for each outlier varied. Some variation was attributed to sample preparation, while others were due to instrument configuration differences. However, having a sound understanding of the effects of the system configuration and the method on the results allowed the sending lab to take a systematic approach to investigating any outliers. Within the study described general steps included:
1. Identifying the outlier results and potential contributions to result
2. Reviewing system configurations, including detector and tubing ID
3. Reviewing sample and mobile phase preparations procedures analyst-to-analyst
This approach can also be used to investigate any out of spec results. This study showed that understanding the risk of the method and implementing control strategies can provide a successful method transfer.
References
- Dlugasch A, Hong P, Tran P. Successful Global Cross Lab Method Transfer of a USP Organic Impurities Method to an Arc HPLC Using a Risk-based Approach. Waters Application Note, 720007285EN, 2021.
- USP, Quetiapine Fumarate. United States Pharmacopeia and National Formulary (USP 43-NF38) 2020, (GUID-DBEED03E-7C75-4167-BD21-4E30BA2EFF2B_2_en-US), 3800.
720007376, September 2021