Global Cross Lab Method Transfer of a USP Impurities Method on the Alliance™ iS HPLC System
Abstract
Implementing and maintaining a competitive advantage is an ever-present challenge in a global economy. For analytical laboratories, this can be accomplished by achieving improvements in both performance and usability through the use of modern High Performance Liquid Chromatography (HPLC) instrumentation. Assessment of a new HPLC instrumentation platform to understand the various system configurations is important to achieving successful results. Another key aspect to reduce the variability from one laboratory to another is to understand specific method conditions and other variables that can impact chromatographic reproducibility. To address these challenges, a solid understanding of variables that can impact method performance should be assessed. Afterward, control strategies to minimize the risks can be implemented. In this application note, a global interlaboratory method transfer of a USP organic impurities method was conducted across eight sites with the Alliance iS HPLC System. The method transfer process was multi-faceted in which control strategies were developed from a risk-assessment that delivered a standard operating procedure (SOP) from the sending laboratory site to the receiving laboratories. With these control strategies, the system and method variables were controlled resulting in a successful global method transfer.
Benefits
- A global interlaboratory study using the Alliance iS HPLC System is feasible when transferring the USP Quetiapine Fumarate Organic Impurities method
- The established configuration of the Alliance iS HPLC System supports a successful method transfer in eliminating configuration variables
- The Alliance iS HPLC System features provide users a way to systematically setup the system by using the established workflows provided
Introduction
In a global economy there is often a challenge to implement and maintain a competitive advantage. New instrumentation is desired as it may provide improvements in both performance and usability. When assessing new instrumentation, with respect to method transfer, it is important to control and understand system configurations. Furthermore, specific method conditions and other variables will need to be controlled to minimize variability from one laboratory to another. To address these challenges, a solid understanding of risks that can impact method performance and control strategies to minimize the risks need to be implemented. Within this application note we will demonstrate that a global interlaboratory method transfer of a USP organic impurities method was conducted across eight sites. Prior to the study, the sending laboratory site conducted robustness and verification testing on the USP quetiapine fumarate impurities method. The process was multi-faceted and included a risk assessment at the sending laboratory site. The control strategies were providing both a standard operating procedure (SOP) and key materials from the sending laboratory site to the receiving laboratories. With these control strategies, the Alliance iS HPLC System, and method variables were controlled to allow for successful method transfer.
The global method transfer was performed on the modernized Alliance iS HPLC System which is a streamlined system with features that help to ensure accurate, reproducible system performance. One example of such features is the modernized interface that enhances the ease of use for HPLC users to systematically and reliably setup the system using the established workflows provided. Implementing a systematic startup each system is setup and equilibrated appropriately and consistently before an analysis.
Experimental
The method utilized was based on the USP method for Quetiapine Fumarate Organic Impurities, with no adjustments.1
Sample Description
The method requires both a system suitability reference standard (RS) and a standard quetiapine fumarate RS. The system suitability solution was prepared from the USP quetiapine system suitability RS (USP p/n: 1592715) and consists of a mixture of quetiapine, quetiapine desethoxy (1–5%), related compound G, and related compound B. The system suitability solution was prepared at 1 mg/mL in diluent (86:14 Solution A/Solution B) from the quetiapine system suitability RS. The standard solution was prepared utilizing the USP quetiapine fumarate RS (USP p/n: 1592704) and was prepared at a concentration of 0.001 mg/mL in diluent. The drug substance was obtained from Hangzhou Think Chemical Co., Ltd. and past the date of expiration. The sample was prepared at 1.0 mg/mL in Solution A.
Method Conditions
|
System: |
Alliance iS HPLC System |
|
|
Detection: |
TUV Detector |
|
|
Configuration: |
Passive Preheater |
|
|
Flow cell: |
Analytical |
LC Conditions
|
Column: |
XBridge™ C₈ 3.5 µm, 4.6 x 150 mm (Waters™ p/n: 186003055) |
|
Column temperature: |
45 °C |
|
Sample temperature: |
10 °C |
|
Injection volume: |
20 µL |
|
UV wavelength: |
250 nm |
|
Data rate: |
10 Hz |
|
Flow rate: |
1.5 mL/min |
|
Run time: |
70 minutes |
|
Buffer: |
3.1 g/L Ammonium Acetate in water. Add 2 mL of 25% ammonium hydroxide to each 1 L of solution, pH is NLT 9.2 |
|
Mobile phase A: |
25:75 Acetonitrile:Buffer |
|
Mobile phase B: |
Acetonitrile |
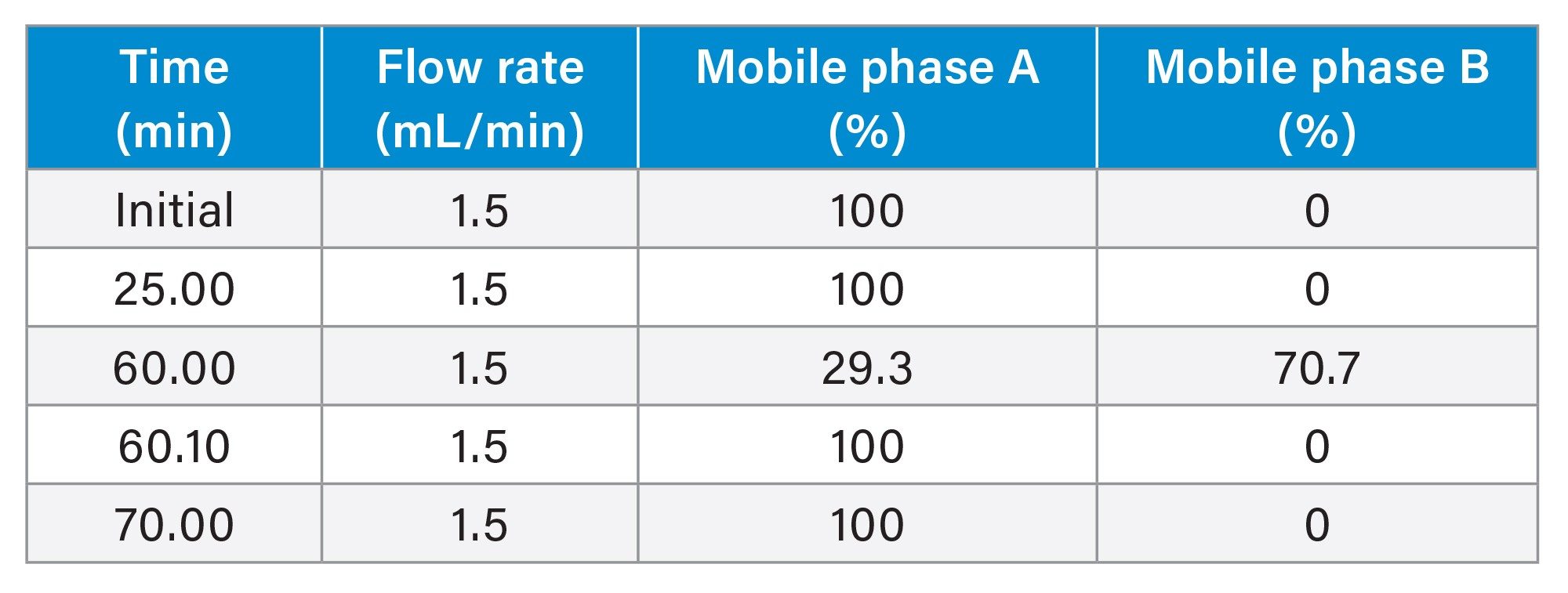
Gradient Table
Data Mangement
|
Chromatography software: |
Empower™ 3 Chromatography Data Software |
Results and Discussion
The method transfer of the USP Quetiapine Fumarate Impurities method was performed across eight sites across three continents. The laboratories were located in Milford, MA, USA (sending laboratory), and seven receiving labs – Texas (USA), Illinois (USA), Indiana (USA), Canada, India, Belgium, and France. The study included verifying the system suitability requirements and quantitative analysis of a drug substance. All analyses were performed on the Alliance iS HPLC System with a Tunable Wavelength (TUV) Detector in which each laboratory received all standards, samples, and columns, wherein there was a total of four different column lots, from the Milford, MA laboratory. Each analysis was assessed using the system suitability specifications as described in the USP method, as well as analyzing a drug substance sample and comparing the impurity results. The system suitability specifications are centered on the resolution of two critical pairs in the system suitability solution, and the tailing, retention time %RSDs and area %RSDs from the standard solution.
To ensure method performance was not impacted by numerous variables, a risk-based approach was completed for the method transfer. This approach consisted of multiple steps including a risk-assessment of the USP method, ranking the risks, and implementing control strategies to control the method variability. These steps and outcomes are described in the previous global cross lab study conducted on the ACQUITY™ Arc™ HPLC System.2 The same control strategies were applied to the cross-lab study on the Alliance iS HPLC System. The SOP generated in the previous study was adjusted to accommodate the new System Startup functionality of the Alliance iS HPLC System. With this new feature of the system, the procedure was standardized to ensure proper setup of each of the Alliance iS Systems, including priming and equilibration before the start of the analysis. Controlling the system startup removes a variable from the method transfer. Through the touchscreen interface the start up procedure simplified this workflow for the analysis.
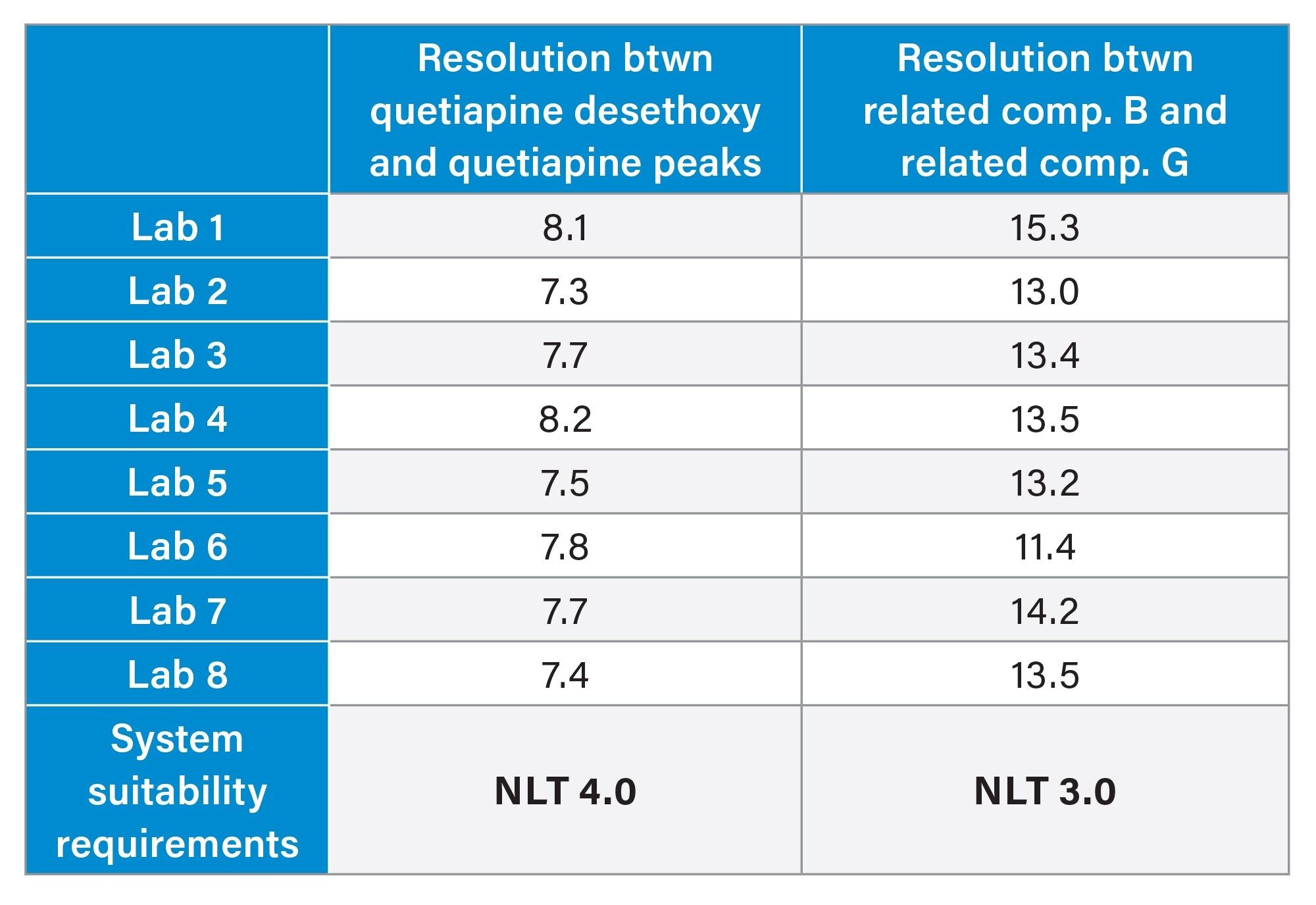
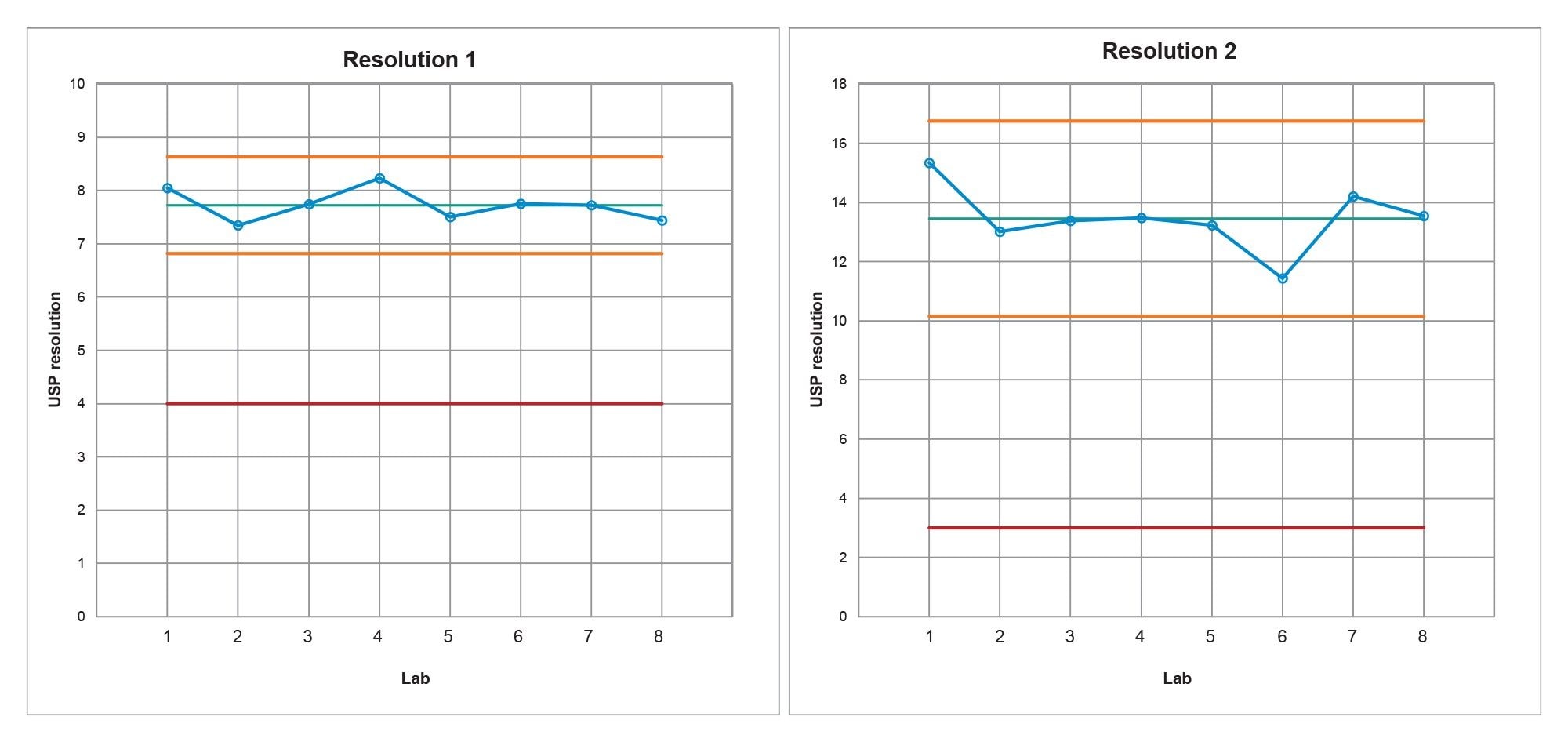
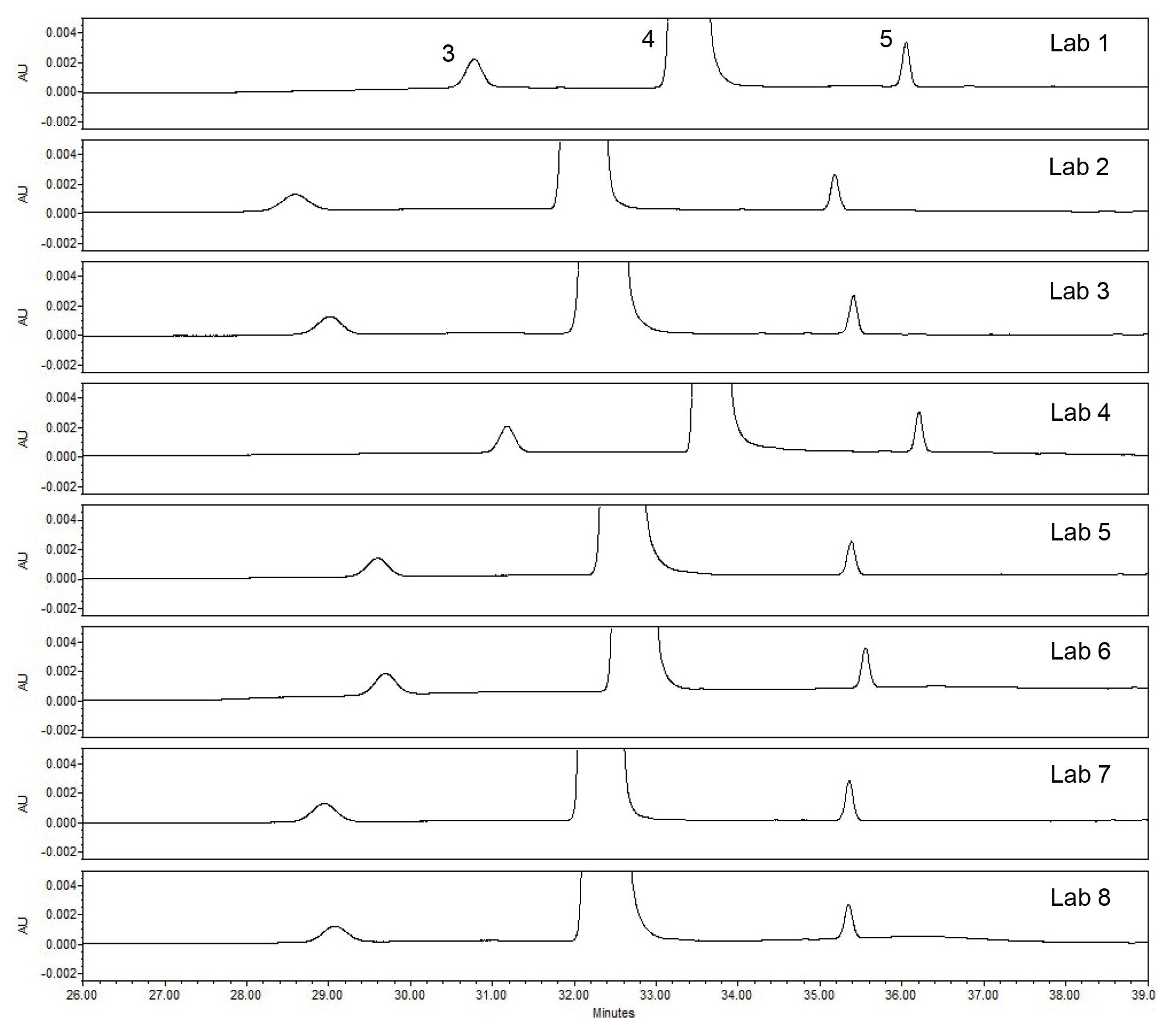
The first step in determining if the method transfer was successful was to assess the system suitability criteria of the USP Impurities method. Within the system suitability criteria, the resolution of two critical pairs is evaluated (Figure 1). The first critical pair (Resolution 1) is between Peak 3, quetiapine desethoxy, and Peak 4, quetiapine. The second critical pair (Resolution 2) is between Peak 1, Related Compound G, and Peak 2, Related Compound B. The results for the resolution between the two critical pairs from all eight laboratories readily met the system suitability requirements (Table 1). To ensure the results are in a state of control, the resolution values obtained from each laboratory were placed into a control chart. The control charts (Figure 2) show the values for Resolution 1 and Resolution 2 are all within the plus or minus three standard deviations of the mean and within the system suitability requirements of the method.
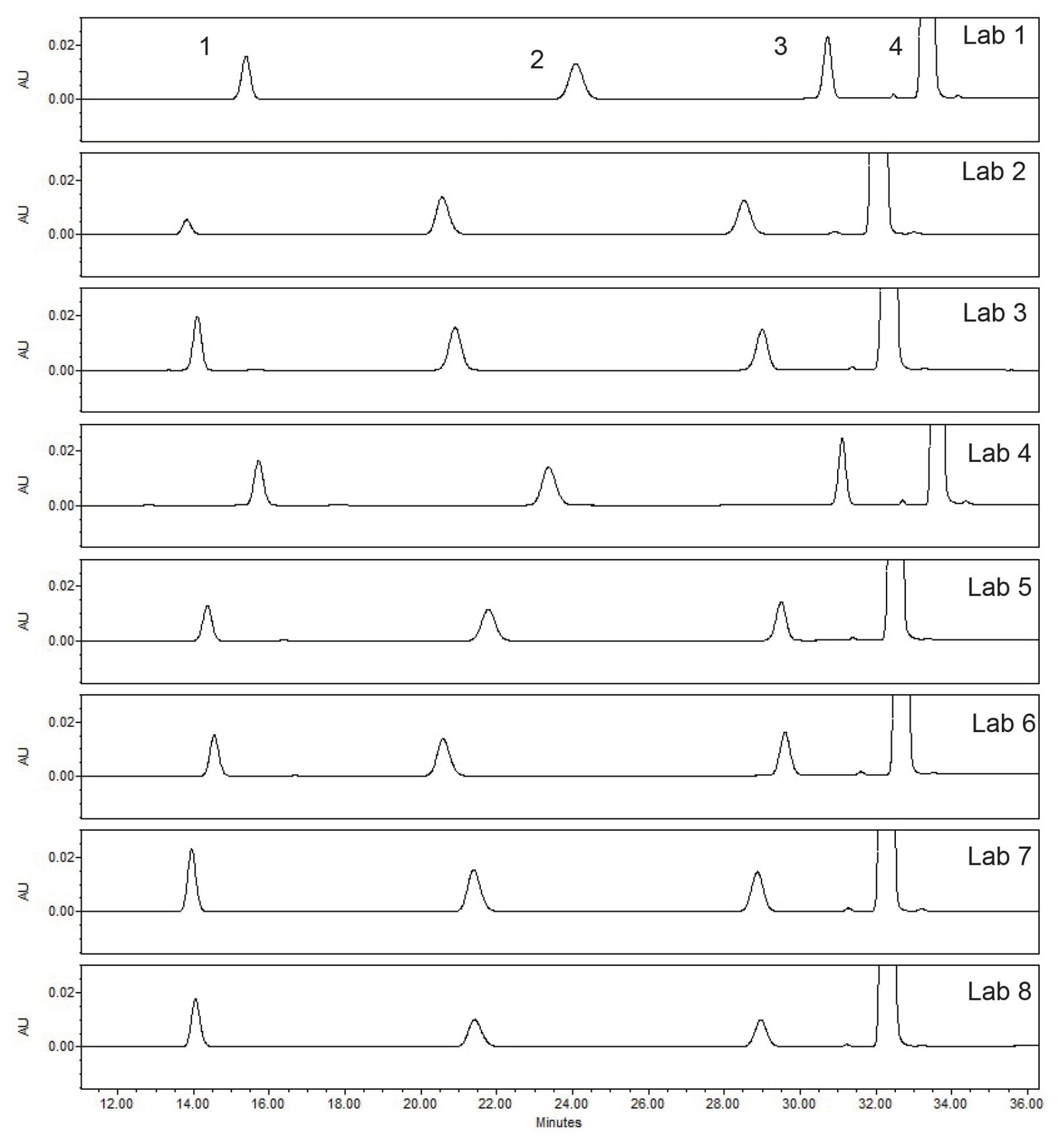
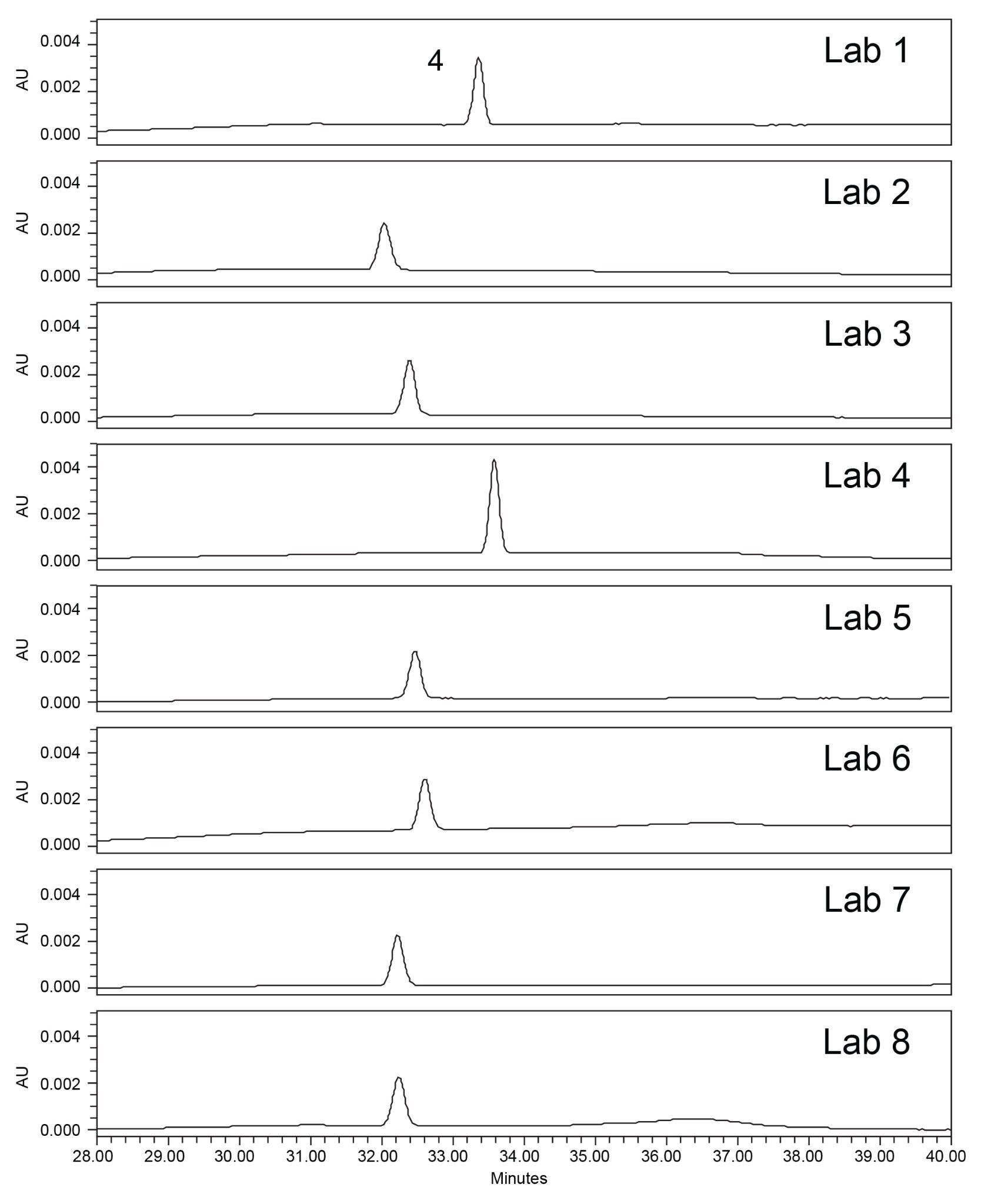
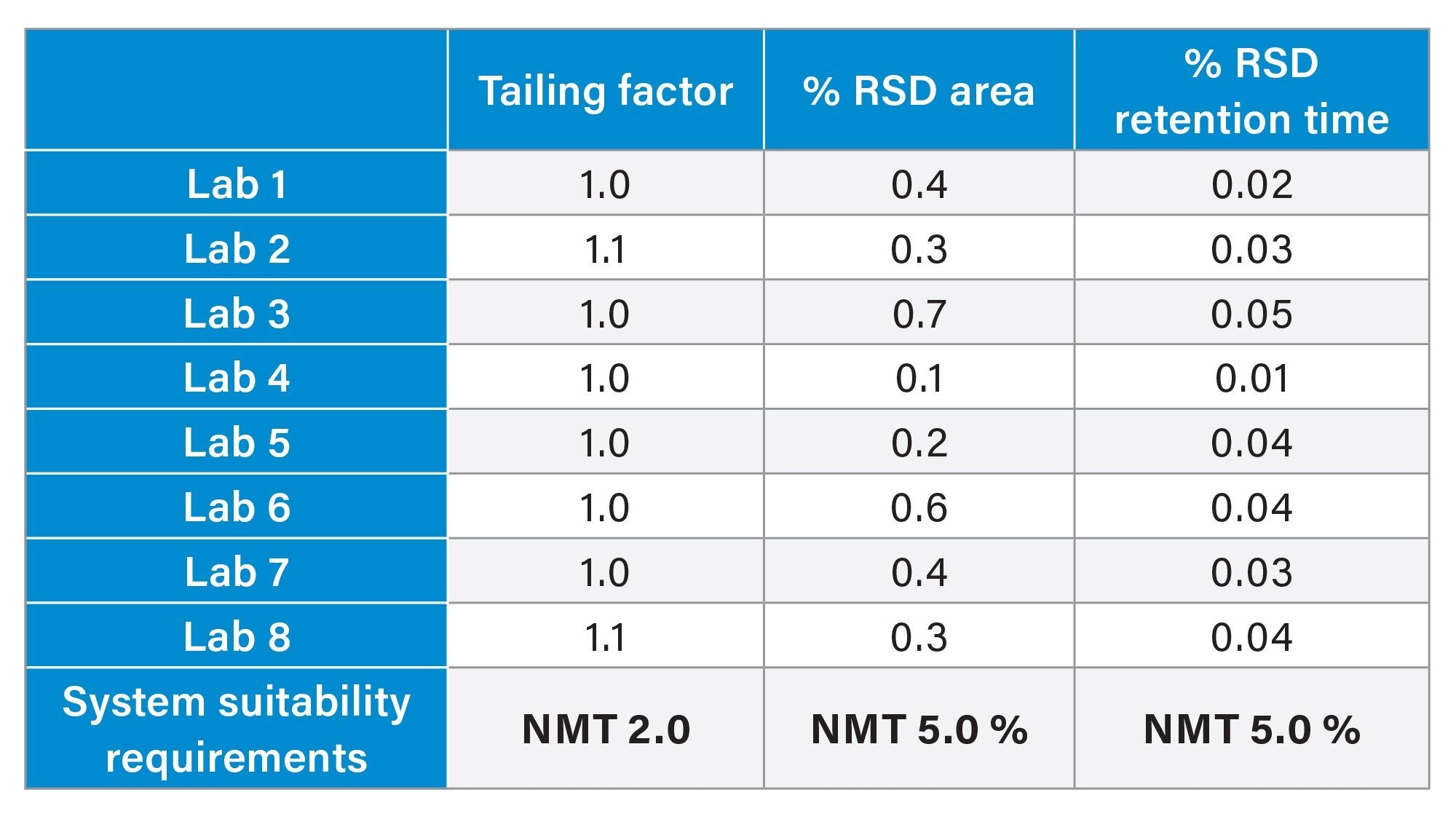
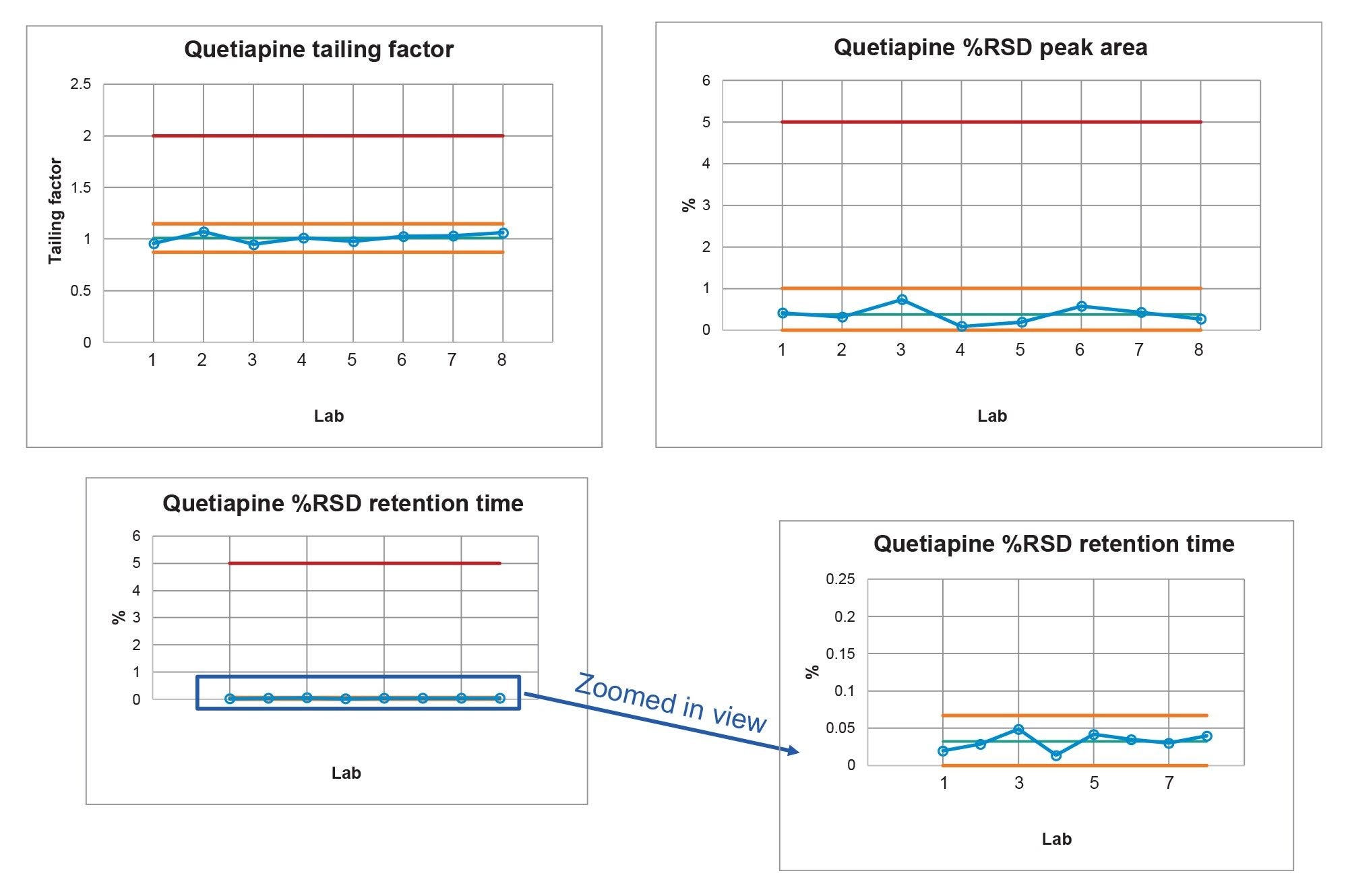
The system suitability requirements of the standard solution were analyzed for tailing factor, retention time %RSD, and area %RSD. The chromatographic results from all eight laboratories (Figure 3) show excellent inter-lab reproducibility on the Alliance iS HPLC System. The results from all eight laboratories (Table 2) meet the system suitability specifications of not more than (NMT) 2.0 for tailing factor, and NMT 5.0% for both area and retention time RSD. All data was placed into control charts (Figure 4) and all data for tailing factor, Area %RSD, and Retention Time %RSD are all within the plus or minus three standard deviations of the mean and within the system suitability requirements of the method.
When comparing the retention times of the compounds for the standard solution and the system suitability solution chromatograms, it is observed from the chromatograms that some of the results from the labs in the study have a slight shift in retention times of the compounds. This retention time shift occurrence is a result of the mobile phase preparation can have a significant impact on the reproducibility of the method. While baselining the method in the Milford, MA laboratory, an analyst had performed robustness studies looking specifically at the ACN/Buffer ratio in the mobile phase and how it could impact the retention time and system suitability. In these studies, the ACN/Buffer ratio was found to have a dramatic impact on retention time in which we did see during the study the previous study as well.3 This minor shift in retention time that we see in this cross-lab study with the Alliance iS HPLC System did not impact the results of the study since all the laboratory sites met the USP system suitability criteria for the system suitability solution and the standard solution.
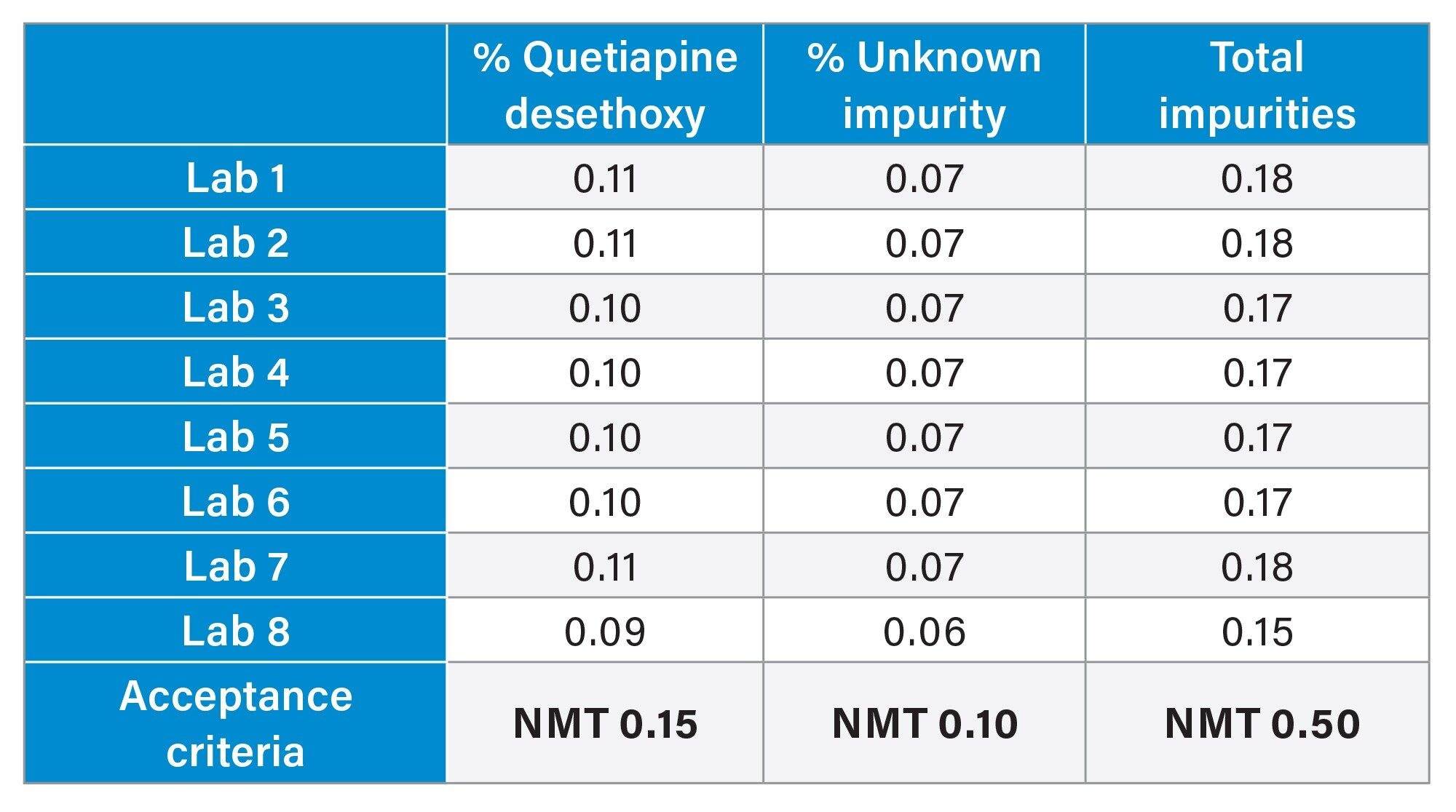
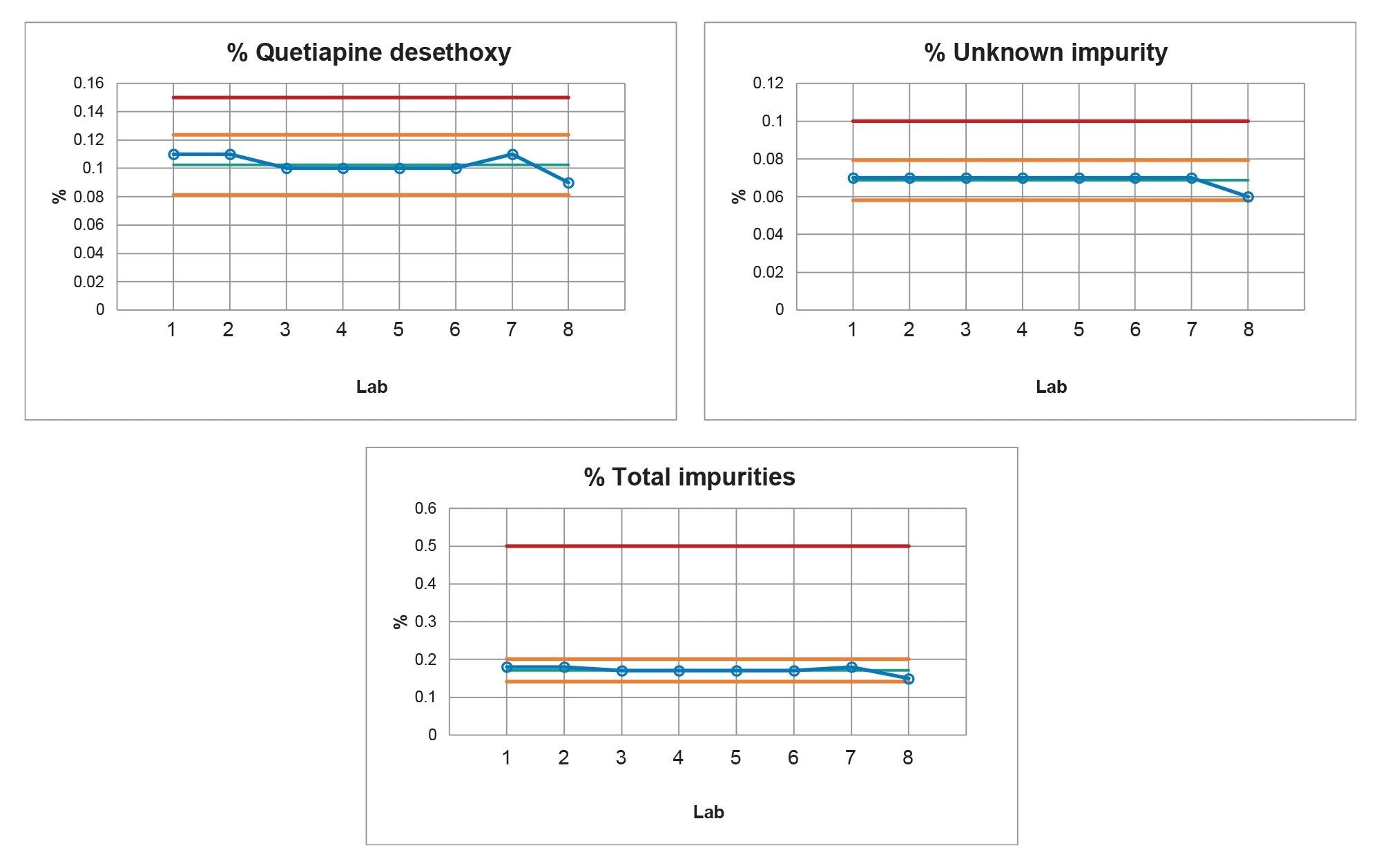
After all eight Alliance iS HPLC Systems met the system suitability specifications, the drug substance was analyzed for the presence of impurities. The analysis found that the drug substance contained two impurity peaks, quetiapine desethoxy (verified by the standard) and an unknown peak that eluted after the main API peak. The drug substance (Figure 5) show reproducible sample results from the Alliance iS HPLC System with the retention times of the two impurity peaks within the expected variation of plus or minus three standard deviations from the mean. The quantitative results from the drug substance sample (Table 3) were within the acceptance criteria of not more than 0.15% for the quetiapine desethoxy, and not more than 0.10% for the unknown impurity. The drug substance that was used for the study was past the expiration date, therefore the presence of an impurity was not unexpected. The control charts (Figure 6), display that the quantitative results were within plus or minus three standard deviations, indicating a state of control. Furthermore, the control limits were less than or equal to the limit specified in the USP method, giving greater confidence that the drug substance met the criteria.
Conclusion
Using a risk-based approach for method transfer across laboratories around the world allowed for a successful transfer of the USP quetiapine fumarate impurities method on the Alliance iS HPLC System. The feature of the modernized interface on the Alliance iS HPLC System provides consistent startup and equilibration with automated workflows and equilibration timing allowing user variability to be controlled. Through this interlaboratory study we were able to demonstrate the ability to successfully replicate the USP quetiapine fumarate impurities method globally on the Alliance iS HPLC System.
References
- USP, Quetiapine Fumarate. United States Pharmacopeia and National Formulary (USP 43–NF38) 2020, (GUID-DBEED03E-7C75-4167-BD21-4E30BA2EFF2B_2_en-US), 3800.
- Dlugasch A, Hong P, Tran P. Successful Global Cross Lab Method Transfer of a USP Organic Impurities Method to an Arc HPLC Using a Risk-based Approach. Waters Application Note. 720007285. 2021.
- Dlugasch A, Hong P, Tran P. Case Study: Investigating Unexpected Results of a Global Cross-Laboratory Study of a USP Organic Impurities Method on an Arc HPLC System. Waters Application Note. 720007376. 2021.
Acknowledgement
Contributors: Jennifer Simeone, Don Tirinite, Larry Meeker, Rosana Jimenez, Cody Kroft, Nadezjna Vaes, Marie-Christine Tang, Padmakar Wagh, Veeranjaneyulu Damisetti
720008321, May 2024